A Review on Cancer, Cause of Cancer and Treatment: Role of PIK3 Pathway on Cancer
The central function of phosphoinositide 3- kinase (PI3K) activation in tumour cell biology has urged a sizeable trouble to target PI3K and/ or downstream kinases similar as AKT and mammalian target of rapamycin (mTOR) in cancer. still, arising clinical data show limited single- agent exertion of impediments targeting PI3K, AKT or mTOR at permitted boluses. One exception is the response to PI3Kδ impediments in habitual lymphocytic leukaemia, where a combination of cell- natural and- foreign conditioning drive efficacity. Then, we review crucial challenges and openings for the clinical development of impediments targeting the PI3K – AKT – mTOR pathway. Through a lesser focus on patient selection, increased understanding of vulnerable modulation and strategic operation of rational combinations, it should be possible to realize the eventuality of this promising class of targeted anticancer agents.
Introduction of Cancer
Cancer is a notable, deadly disease that is large, complex, and characterized by unchecked growth and spread of abnormal cells. These abnormal cells can be aggressive (develop and divide outside of normal limits), invasive (invade and damage nearby tissues), or metastatic (spread to other parts of the body). These malignant characteristics of cancer set them apart from benign tumors, which do not penetrate or spread, and whose growth is self-contained. Cancer is brought on by both internal (inherited mutations, hormones, immunological conditions, and metabolic mutations) and external (tobacco, infectious organisms, chemicals, and radiation) causes. Although it can afflict anybody, including fetuses, the risk factor for its common forms tends to rise with age and weight [1].
Chemotherapy, radiation therapy, surgery, monoclonal antibody therapy, immunotherapy, and other therapies are all options for the treatment of cancer. The preferred course of treatment is determined by the patient’s overall health, the location, grade, and stage of the disease, as well as the tumor. There is an urgent need for the creation and development of innovative anticancer medications that could successfully suppress proliferative pathways. In more advanced eukaryotes, the physiological process of cell death known as apoptosis functions as a defensive mechanism and plays a crucial role in preventing the development of tumor cells [2]. Numerous anticancer drugs cause tumor cells to undergo the apoptotic process, which is thought to be substantially responsible for its dysregulation in the pathogenesis of cancer [3]. Major problems with cancer treatment include cytotoxicity and genotoxicity of anticancer medications to normal cells, which raises the possibility of causing subsequent malignancy [4]. Therefore, there has been a lot of focus on developing medications with low cytotoxicity that can efficiently induce death in tumor cells.
Every country, region, and socioeconomic level are affected by the significant global health burden that is cancer. Today, cancer is responsible for more deaths than HIV/AIDS, TB, and malaria combined, accounting for one out of every eight worldwide. One of the most challenging conditions for chemists and oncologists is cancer treatment that targets tumor growth. According to figures on cancer fatalities worldwide given by the American Cancer Society, there were 7.6 million cancer-related deaths worldwide in 2007, or nearly 20,000 per day, with 38% occurring in affluent nations and 62% in developing ones. Additionally, the burden of cancer worldwide is increasing at a startling rate. It is projected that by 2050, 27 million new cancer cases will be registered and 17.5 million deaths may also be caused to occur in the world [5].
World Wide Cancer Condition
The number of new cancer cases and cancer-related deaths among AYAs was estimated to be 1 335 100 worldwide [95% uncertainty interval (UI) 1 243 397-1 426 785] and 397 583 [95% UI 371 460-426 061], respectively, with a female: male ratio of 1.35 for incidence and 1.04 for mortality. In 2019, the incidence rate of cancer was 44.99 and the death rate was 13.39 per 100,000 persons.
The cancer burden among AYAs was characterized by a predominance of females. Females had a 52.32 versus 37.82 per 100 000 incidence rate for all malignancies, which was 38.3% higher than that of males. The 29 different cancer kinds carried quite different burdens. Leukemia (1.51/100 000), breast cancer (1.45/100 000), brain and CNS cancer (0.98/100 000), colorectal cancer (CRC) (0.95/100 000), and stomach cancer (0.93/100 000) rounded out the top five cancer types overall in terms of new cases. The top four were nonmelanoma skin cancer (4.74/100 000), cervical cancer (4.01/100 000), leukemia (3.41/100 000), and colorectal cancer (2. Just below lung cancer, which accounts for 18% of cancer fatalities in 2020, CRC accounts for 10% of global cancer incidence and 9.4% of cancer deaths. According to projections of population increase, aging, and human progress, there will be 3.2 million additional CRC cases worldwide by 2040. The rise in CRC incidence is mostly due to increased exposure to environmental risk factors brought on by a westernization of lifestyle and nutrition [6, 7, 8, 9].
Globally, there will be 50,000 new cases of anal cancer, 0.7 million new cases of rectal cancer, and 1.15 million new cases of colon cancer in 2020, according to estimations from GLOBOCAN 2020 [10, 11]. These figures are expected to rise to 1.92 million, 1.16 million, and 78,000 in 2040, respectively, with further growth anticipated, as shown in Table 1.
| Cancer Sites | 2020 | 2040 |
|---|---|---|
| Colon | 1,148,515 | 1,916,781 |
| Rectum | 732,210 | 1,160,296 |
| Anus | 50,865 | 77,597 |
| Total | 1,931,590 | 3,154, 674 |
Table 1: Estimated number of new cases from 2020 to 2040.
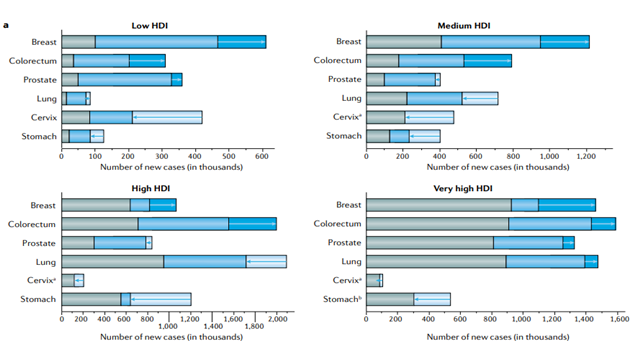
We calculated the number of cancer cases in 2018 along with the anticipated increases until 2070 as a result of demographic changes (population growth and aging) and changes in the underlying rates as a result of changes in risk factors that either cause an increasing or decreasing number of predicted cancers (Figure 1). We project 9.1 million new instances of breast and colorectal cancer in 2070, up 131% from 2018, as a result of demographic shifts and growing incidence. As a result, a drop in the number of cancer cases is predicted: 409,000 instances in 2070 versus 484,000 in 2018 [12]. In contrast, the reported decline in the incidence of cervical cancer in nations with medium-to-high HDI negates the demographic effect. In general, the advancements in cancer control that have resulted in decreasing incidence for other cancer types (including cervical and stomach cancers, as well as lung cancer in males) cancel out the growing incidence of some cancer types (breast, colorectal, and prostate) [13].
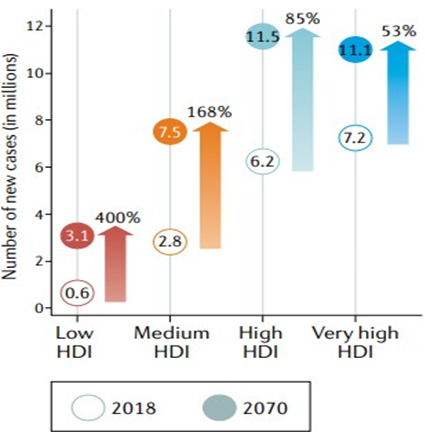
In 2070, there will likely be twice as many new cases of cancer worldwide—34 million—as there were in 2018. Countries with a high HDI are expected to see the biggest absolute increases, closely followed by those with a medium HDI, increasing from 6.2 million and 2.8 million cases, respectively, in 2018 to 11.5 million and 7.5 million cases in 2070 (Figure 2) [14] .
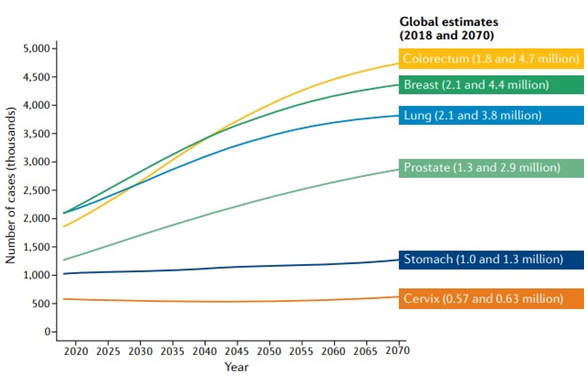
According to predictions, colorectal and breast cancer cases will rise from 1.8 million and 2.1 million, respectively, in 2018 to 4.7 million and 4.4 million, respectively, yearly by 2070 (Figure 3) [15].
Targeting Pathway
Cancer remains a major concern on a global scale despite the huge efforts that have been made to deploy novel chemotherapeutic techniques for the treatment of cancer. Therefore, it is vital to look for additional therapeutic drugs that have a particular effect against different cancer cells [16]. The phosphatidylinositol 3-kinase (also known as phosphoinositide 3-kinase, PI3K)/Akt (protein kinase B) and the mammalian target of rapamycin (mTOR) signaling pathways are critical in pathological and physiological settings for many aspects of cell growth and survival. Because of their strong connections, they are frequently viewed as one single, distinct pathway that interacts with a number of other pathways, including Notch, Wnt, c-Jun N-terminal kinase (JNK), Ras, Hypoxia Inducible Factors (HIFs), Mitogen- Activated Protein Kinase (MAPK) [17, 18, 19, 20].
PI3K-Akt-mTOR Signaling Pathway: An Overview
For cells to survive and develop in pathological circumstances like cancer, the PI3K-Akt-mTOR pathway is essential [21]. In the physiological response to external stimuli including insulin, insulin-like growth factor-1 (IGF-1), fibroblast growth factor (FGF), and epidermal growth factor (EGF), the PI3K-Akt-mTOR pathway is a key signaling hinge [22]. Some notable upstream regulators of the PI3K-Akt pathway include chemokine receptor-9, toll-like receptors (TLRs), which are associated with inflammatory cytokines, and interleukin-6 (IL-6) [23, 24, 25].
Class I PI3Ks
The catalytic domain of the first class of PI3Ks, class I, is attached to a regulatory subunit or adaptor before being activated by the cell surface receptor, which brings them into close contact to their substrates [26]. Tyrosine kinase (RTKs) receptors, Ras, and G-protein-coupled receptors all have a role in the activation of class IA of PI3Ks. Class IB PI3Ks, on the other hand, can only be triggered by G-protein-coupled receptors. Class I PI3Ks are involved in cell growth, survival, and proliferation more generally [27].
Class II PI3Ks
Class II PI3Ks must engage with regulatory subunits, but they can also interact with other possible adaptor proteins. They can be triggered by cytokine receptors, RTKs, and integrins and contain a Ras-binding motif as well. PIP2s and phosphatidylinositol (PI) serve as their sources of energy. Class II PI3Ks may have a function at the cell cortex; they are also thought to influence insulin signaling, glucose transport, cell migration, membrane trafficking control, and receptor internalization [28].
Class III PI3Ks
There is currently just one class III PI3K known, called Vps34, which uses PI as a substrate. It is thought to regulate a number of membrane trafficking processes, including cytokinesis, autophagy flux, endosome-lysosome maturation, and endosomal protein sorting. In addition to being involved in cell growth and survival, downstream targets are also controlled in response to cellular stress and the supply of amino acids [29, 30]. The precise regulatory framework is provided in Figure 4.
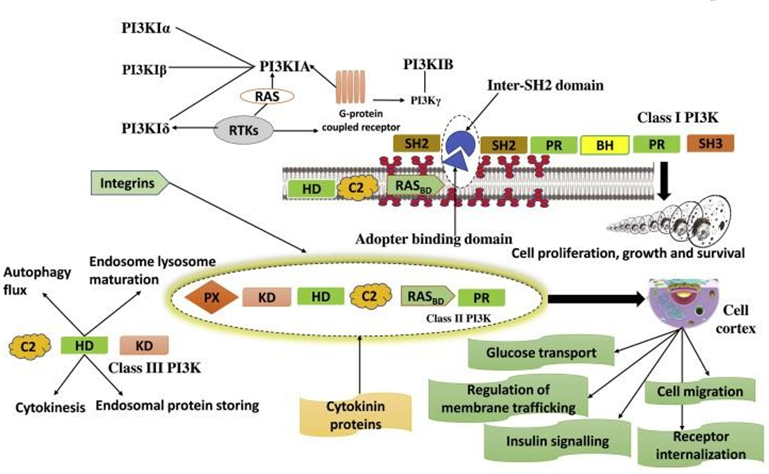
The class IA of PI3Ks activates with the help of tyrosine kinase (RTKs) receptors, either directly or via Ras and through G-protein-coupled receptors. Class II of PI3Ks activates through cytokine receptors, RTKs, and integrins. Class III PI3K is responsible to regulate various membrane trafficking functions, including endosome-lysosome maturation, endosomal protein sorting, autophagy flux, and cytokinesis.
Activation of the PI3K-Akt-mTOR Pathway
Amplification of mutations that impact different PI3K-Akt pathway proteins or their activation can both result in PI3K-Akt pathway activation. These include the inactivation of mutations or deletions in phosphatases that cause hydrolysis of PI3K products, like phosphatidylinositol 3,4,5-trisphosphate (p1p3), PTEN, and tumor suppressors INPP4B and PIK3R1, the adaptor protein, and PIK3CA (p110a), the type I PI3K isoform [31].
PI3K-Akt-mTOR Signaling and Cancer
Due to the critical function of the PI3K-Akt-mTOR pathways in cellular functioning and pathologic change, malignancies frequently have mutations linked to this system [32]. Cancer development has been strongly linked to genetic alteration that targets the PI3K-Akt-mTOR pathway. For example, somatic loss of PTEN due to deletion or genetic mutation is the second most frequent oncogenic mutation in the human genome, overshadowed by p53 malignant alterations [33].
The impact on lung cancer patients and the connection between mTOR and interleukin-7 (IL-7) in human lung cancer cells were demonstrated. They discovered that the PI3K-Akt-mTOR signaling pathway was activated by IL-7, which also increased PI3K expression and encouraged Akt and mTOR phosphorylation. Breast cancer cell lines, Pre-B leukemia, and T cells have all been shown to activate the PI3K-Akt-mTOR signaling pathway in response to IL-7 [34].
Investigated the connection between the PI3K-Akt/ mTOR pathway and the HOXD antisense growth-associated long non-coding RNA (HAGLROS). In colorectal cancer cells, they discovered that overexpressing HAGLROS alone significantly lowers PTEN expression levels and noticeably raises the expression of pAkt, pPI3K, and pmTOR. Additionally, it was discovered that the PI3K-Akt-mTOR pathway was solely triggered by the overexpression of HAGLROS [35]. The influence of positive and negative growth regulators on PI3KAkt-mTOR is shown in Figure 5.
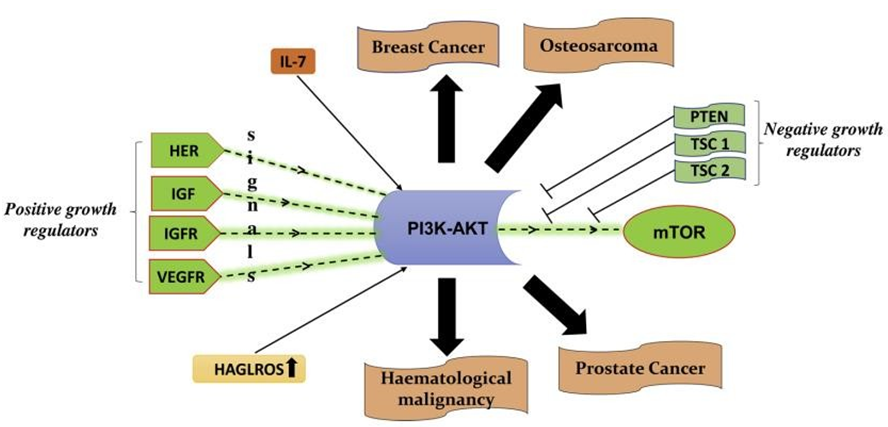
Figure 5: Positive and negative growth regulators of PI3K-Akt. IL-7 upregulated PI3K expression and promoted Akt- mTOR phosphorylation for the activation of the PI3K-Akt -mTOR signaling pathway. The HOXD antisense growth- associated long noncoding RNA (HAGLROS) overexpression alone can reduce the PTEN expression levels and HAGLROS overexpression alone can activate the PI3K-Akt-mTOR pathway.
PI3K-Akt-mTOR Signaling Pathway in Prostate Cancer
One of the most notable health issues for males is prostate cancer. Over the past few decades, prostate cancer deaths have soared. Furthermore, it was noted that the 256,000 deaths that were reported in 2010 marked a significant increase from the 156,000 deaths that were reported in 1990 [36]. As a major therapeutic target and a very practical option for a biomarker that predicts the genesis, progression, and behavior of prostate cancer following prostatectomy, Torrealba and colleagues suggested the PI3K-Akt-mTOR pathway [37].
PI3K-Akt-mTOR Signaling Pathway in Breast Cancer
The management of breast cancer has undergone a remarkable transformation as a result of early detection. Women who have small primary tumors and negative axillary lymph node dissections (stage I of the disease) are currently typically treated with surgery, which has an 80% recovery rate [36]. Because of their capacity to briefly switch between mesenchymal and epithelial states, breast cancer cells have the potential to spread throughout the body. The PI3K-AktmTOR pathway is frequently activated in breast cancer, and PIK3CA is the most frequently mutated gene of significant relevance in ER-positive breast cancer [37].
PI3K-Akt-mTOR Signaling Pathway in Osteosarcoma
Osteosarcoma is one of the most prevalent malignant bone tumors with lung metastases and a recurrent disease in people [38]. Inhibition of the PI3K-Akt signaling pathway results in enlarged apoptotic cells in osteosarcoma by downregulating the inhibitor of caspase-9, caspase-3, and apoptosis protein activation [39]. As a result, targeting the PI3K-Akt pathway has received a lot of interest lately for the creation of anticancer drugs [40]. For the prevention and treatment of osteosarcoma, targeting the mTOR signaling pathway has the potential to be a highly effective therapeutic strategy [41].
PI3K-Akt-mTOR Signaling Pathway In Hematological Malignancies
The PI3K inhibitors are a crucial addition to leukemia and lymphoma therapy, but there are now only two licensed PI3K inhibitors due to availability, which raises the possibility of fatal and major adverse events [42].
A poor outcome in hematologic malignancies is linked to frequent deregulation of the PI3KAkt-mTOR signaling pathway. In around 50–80% of cases of acute myeloid leukemia [43, 44] and in roughly 87.5% of cases of T cell–acute lymphoblastic leukemia (T-ALL), the activation of the PI3K– Akt–mTOR signaling pathway was found to be beneficial. Chronic lymphocytic leukemia (CLL), chronic myelocytic leukemia (CML), high-risk myelodysplastic syndrome (MDS), and multiple myeloma (MM) all exhibit activation of the PI3K-Akt-mTOR pathway [45].
Cause of Cancer
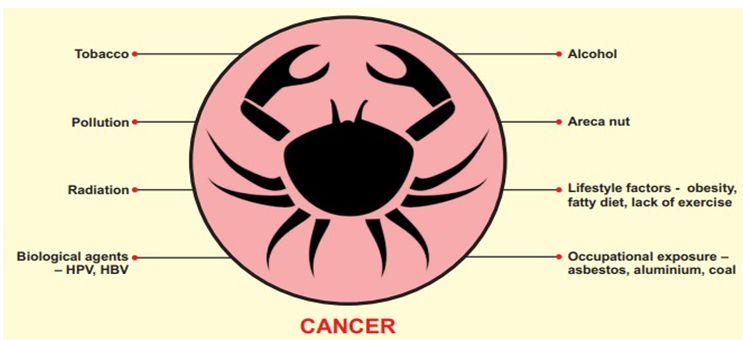
• There are a variety of factors that can result in cancer in various body parts, including the use of tobacco (22% of deaths), poor diet, obesity, inactivity, excessive alcohol consumption (or other factors), and exposure to ionizing radiation, environmental pollutants, and infection [46, 47, 48].
• Hepatitis b, hepatitis c, human papillomavirus infection, helicobacter pylori, immunodeficiency virus (HIV), and Epstein-Barr virus are some of the infections that can lead to cancer. These elements have altered the genes, at least in part.
• Additionally, 5–10% of cancer cases are caused by inherited genetic flaws from the patient’s parents.
• Three types of external agents genetic factors, environmental factors, and agents we consume interact to produce cancer.
• Physical Carcinogens: Ionizing radiation such as radon, ultraviolet rays from sunlight, uranium, radiation from alpha, gamma, beta, and X-ray-emitting sources.
• Chemical Carcinogens: About 60 known highly strong cancer-causing compounds or substances, including nitrosamines, asbestos, cadmium, benzene, vinyl chloride, nickel, and benzidine, are found in tobacco use or smoking, as well as in drinking water contaminants like arsenic and food contaminants like aflatoxin [46].
• Biological Carcinogens: infections caused by certain types of bacteria, viruses, or parasites, including helicobacter pylori, kaposi’s sarcoma-associated herpesvirus, markel cell polyomavirus, hepatitis B and c, human papillomavirus (HPV), Epstein-Barr virus (EBV), and hepatitis B and C [46].
What are the Common Causes of Cancers
Common causes of cancer are as shown in Figure and are as follows (Figure 6):
Tobacco The single most significant preventable risk factor for cancer death worldwide is tobacco use. It is thought to be responsible for 22% of cancer deaths annually, according to the WHO. The vast majority of lung cancers are linked to smoking. Adults who don’t smoke have also been linked to malignancies by passive smoking.
Risk rises as smoking frequency increases. The usage of smokeless tobacco is more common across the Indian subcontinent. These include using raw tobacco, betel quid, gutkha, pan masala, masheri, and other substances. In addition to some types of leukemia, smoking causes malignancies of the nose, sinuses, cervix, ovary, mouth, throat, oesophagus, urinary bladder, pancreas, kidney, liver, and stomach (Figure 7) [47].
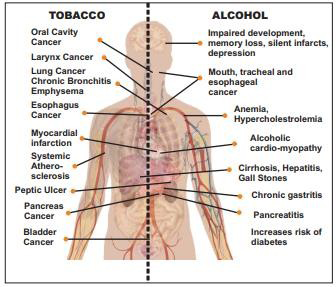
• Tobacco: contains over 4000 types of chemicals. Out of these, around 200 are harmful for human body and about 70 different chemicals have been found to be carcinogenic (Figure 8). About 50% of tobacco users die because of some form of tobacco related disease.

• Alcohol: Alcohol use alone has been linked to a number of malignancies, and the risk grows as alcohol consumption does as well. When combined with smoking, it has a synergistic effect that makes the risk of cancer development considerably higher than when either substance is consumed alone. Alcohol consumption increases the risk of developing a variety of cancers, including those of the breast, liver, colon, rectum, pharynx, larynx, and esophagus.
• Areca Nut: In India, it is also known as supari. When combined with betel leaf, catechu, and slaked lime, it is known as Pan or betel quid and can be chewed alone or in a mixture. Pan Masala is a ready-to-eat blend of areca nut powder and other components.
• Pollution: This includes the environmental pollution of air, water and soil with carcinogenic chemicals. Exposure to these chemicals can occur through air and drinking water. Chemicals like arsenic cause contamination of drinking water and can result in lung cancers.
• Lifestyle factors: These include unhealthy eating habits and lack of physical activity. High body mass index is associated with cancers of esophagus, colorectum, breast, endometrium etc. Excessive consumption of red meat is associated with colorectal cancers.
• Obesity: It refers to excessive fat accumulation. If body mass index (BMI) is above 25 the person is called overweight. Those having BMI over 30 are called obese. It is associated with increased risk of heart diseases, diabetes and cancer like those of endometrium, colon, breast, esophagus, pancreas etc.
• Occupational exposure: Several cancers are seen to have higher association with specific occupations. Substances like asbestos, cadmium, ethylene oxide, benzopyrene, silica, ionizing radiation including radon, tanning devices, aluminum and coal production, iron and steel founding, have been found to be associated with various cancers.
• Radiation including UV rays: These include various viruses, parasites and bacterial infections which predispose the patient to develop certain cancers. Human papilloma virus (HPV) is associated with cervical cancers and oropharyngeal cancers. Hepatitis B and C are associated with liver cancers.
Treatment of Cancer
Depending on the type of cancer and how far along it is, there are several forms of cancer therapies. Many cancer patients receive a combination of treatments, such as surgery and radiation therapy, rather than just one cancer treatment. Clinical examination: A patient with cancer must have a full history of their symptoms as well as a clinical examination. An effort is made to determine the extent of the disease and how it affects nearby critical structures. Endoscopy may be done if necessary to evaluate the lesion. It is also checked for the presence of any other illnesses that can influence how cancer is treated. Evaluation of any distant metastases present is also performed.
• Radiologic assessment: Radiologic imaging is used in addition to a clinical assessment. Depending on the clinical need, this may also comprise X-rays, Ultrasonography, CT scans, MRI scans, or PET scans. They said in a more accurate assessment of the lesion, its size, and the presence of local or distant metastases.
• Pathologic assessment: A tiny portion of the lesion is biopsied along with the clinical evaluation and sent for histological analysis. If necessary, a sample of the lesion’s aspiration is submitted for cytological analysis. The presence and type of cancer cells are determined in each of these tests using a microscope and a few unique stains. The type and stage of the cancer will determine the next course of treatment.
• Surgery: Surgical excision with reference to cancer refers to excision of the lesion with wide margins. Along withthe primary lesion, regional lymph nodes are also addressed. The defect created may be closed primarily or may have to be reconstructed with local, regional or free flaps (Figure 9).

Radiotherapy: Ionizing radiation is used in the process to treat cancer. It can be applied as a primary modality or as a supplement to other therapies utilized after or before surgery. Ionizing radiation causes DNA damage and cell death by forming free radicals in the body. The radiation beam is designed to harm nearby healthy tissues as little as possible. External beam radiotherapy is the term used when the radiation source is external to the body (Figure 10).
• Chemotherapy: A variety of medications are used to treat cancer, as mentioned in Figure 11. These medications trigger cell death at various stages of the cell cycle through a variety of mechanisms of action. The specifics of their mode of operation and activity are outside the purview of this chapter. These could be applied to treat cancer in various contexts. It may be used as the main treatment method in conjunction with radiation, in an adjuvant setting, or as a palliative therapy.
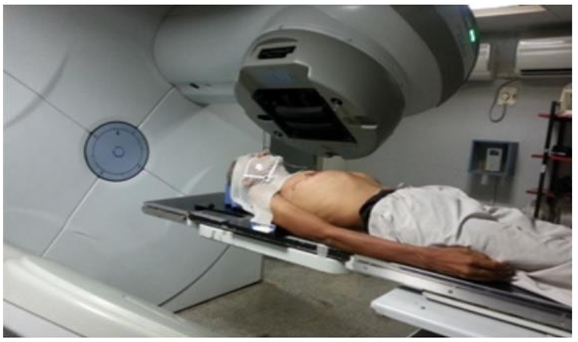
• Palliative care: Many times, cancer can advance past the point at which a curative treatment is still viable. The patient’s disease could be in a very advanced stage or it could have spread to other organs. When a cure is not anticipated, the patient is given palliative care. The goal of palliative care is to manage cancer symptoms rather than treat the disease itself. A chosen subset of patients may receive radiotherapy or chemotherapy. If a patient’s overall health is really poor, optimum supportive care is provided for him.
P13K Pathway
Phosphatidylinositol 3-kinase (PI3K) was first identified more than 20 years ago. Viral oncoproteins’ capacity to transform depended on their interaction with a PI3K lipid kinase activity [48]. In the years that followed, research confirmed the pivotal function of PI3K signaling in a number of cellular functions essential for the development of cancer, such as metabolism, growth, survival, and motility. One of the most common manifestations of PI3K signaling when it is not appropriate is human cancer [49, 50, 51]. As a result, major attempts have been made to create PI3K pathway inhibitors that can be used to treat cancer. However, it is yet unclear which tumors will respond best to these therapies and how to employ them most effectively. The potential therapeutic benefit of PI3K pathway inhibition may also be constrained by the numerous adverse physiologic consequences of PI3K inhibition. Here, we’ll examine the evidence for PI3K’s involvement in the growth and maintenance of tumors. We will contrast the several potential therapeutic approaches for blocking this system and how the mode of PI3K pathway activation in a specific malignancy may affect their success. Finally, we will go over recent research comparing the advantages of using PI3K pathway inhibitors alone versus in conjunction with other drugs to treat cancer [52].
PI3K Signaling Cascade Regulates Cell Growth and Surviva
PI3Ks are divided into three classes based on their structure and function. The most blatantly implicated PI3K subclass in human cancer is Class IA [53]. A regulatory subunit plus a catalytic subunit make up Class IA PI3Ks. P85 (p85, p55, and p50 isoforms), p85, and p55 regulatory subunits are encoded by three mammalian genes, PIK3R1, PIK3R2, and PIK3R3, and are collectively referred to as p85 [54]. Three genes, PIK3CA, PIK3CB, and PIK3CD, produce the catalytic isoforms, p110, p110, and p110. Both PIK3CA and PIK3R1 are somatically mutated in malignancies, and these alterations encourage activation of the PI3K pathway, as will be covered in more depth below [55, 56, 57, 58, 59, 60, 61].
Growth factor stimulation causes receptor tyrosine kinases (RTKs) to activate Class IA PI3Ks. The regulatory component, p85, directly binds to RTK and/or adaptor phosphor-tyrosine residues. This interaction frees the p110 catalytic subunit from the intermolecular inhibition caused by the p85 protein and directs PI3K to the plasma membrane, where its substrate, phosphatidylinositol 4,5-bisphosphate (PI, P2), is found [62]. Ras that has been activated and is binding to p110 directly can also stimulate PI3K. Additionally, G-protein coupled receptors have the ability to activate the p110catalytic subunit.
To create PI [3, 4, 5] P3, PI3K phosphorylates PIP2 at the 3OH location. PIP3 is dephosphorylated to PIP2 by the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which stops PI3K-dependent signaling. By directly interacting with the pleckstrin homology (PH) domains of different signaling proteins, PIP3 spreads intracellular signaling [63]. Phosphoinositide-dependent kinase 1 (PDK1) and AKT are two serine/threonine kinases that possess PH domains, and PI, P3 brings them together. By phosphorylating AKT at threonine 308, PDK1 activates AKT [64]. Through a number of methods, PI3K-AKT signaling supports cell growth and survival. BAD and BAX, two members of the proapoptotic Bcl-2 family, are inhibited by AKT to aid in cell survival [65]. Additionally, AKT prevents NF-B from being negatively regulated, which promotes the transcription of more prosurvival and antiapoptotic genes. AKT negatively regulates for khead transcription factors, lowering the expression of cell death-promoting proteins, and phosphorylates Mdm2 to counteract p53-mediated apoptosis [66]. AKT also phosphorylates TSC2, which prevents the TSC1/TSC2 dimer from acting as a rheb GTPase. Increased p70 S6 kinase activity is brought on by activated rheb stimulating the mTORC1 protein complex, which contains the mTOR gene. By phosphorylating the ribosomal S6 protein and eukaryotic initiation factor 4E, mTORC1 activation increases protein synthesis (Figure 12) [67].
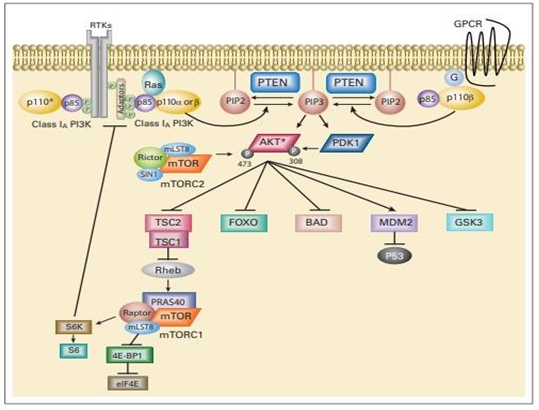
Figure 12: The phosphatidylinositol 3-kinase (PI3K) signaling cascade. PI3K signaling impacts on cell growth, survival, and metabolism. Arrows represent activation, while bars reflect inhibition. A negative feedback loop has been described from the downstream target S6 kinase (S6K) to the adaptor protein IRS-1. RTK, receptor tyrosine kinase; GPCR, G-protein coupled receptor; P, phosphate; G, G protein; PTEN, phosphatase and tensin homolog; IRS-1, insulin receptor substrate 1; eIF4E, eukaryotic initiation factor 4E; S6, ribosomal S6 protein; PIP2, phosphatidylinositol 4,5-bisphosphate; mTORC2, rapamycin (mTOR) – containing protein complex 2. (*) p110 alpha, beta, or delta
Inhibitors of PI3K Signaling in Cancer Treatment
In some cases, PI3K signaling inhibition can increase cell death and reduce cell proliferation. As a result, this pathway’s components make good targets for cancer treatments. Many PI3K pathway inhibitors have been created, and they are now being examined in preclinical research and early clinical trials. The most developed rapamycin analogs in the clinic have been approved for the treatment of advanced renal cell carcinoma by the US Food and Drug Administration, including temsirolimus and everolimus, which specifically inhibit mTORC1 [68]. It will not be examined further because the use of rapamycin analogs in the treatment of cancer has already been thoroughly reviewed elsewhere [69]. The possible therapeutic functions of various PI3K pathway inhibitors will be covered in this review. AKT inhibitors, dual PI3K-mTOR inhibitors that block the p110isoforms of mTOR and the kinase component of both mTORC1 and mTORC2, and PI3K inhibitors (both pan-PI3K and isoform- specific PI3K inhibitors) are a few of these. These drugs may affect tumor angiogenesis, metastasis, and metabolism in addition to their capacity to block cancer cell proliferation and survival signals, as previously mentioned [70].
PI3K and Insulin Signaling: Potential Toxicity and Pharmacodynamic Marker of PI3K Inhibition
Through the course of eurkaryotic evolution, PI3K signaling has played a crucial part in mediating the effects of insulin on cellular metabolism [71]. Reduced PI3K response to insulin signaling is linked to non-insulin-dependent diabetes mellitus, which is characterized by insulin insensitivity [72]. The functional importance of this system on glucose homeostasis has been confirmed by a number of transgenic and knockout animals carrying changes in p85, p110, PTEN, or AKT2. These findings imply that patients using PI3K pathway inhibitors may experience insulin resistance, and this condition may actually be employed as a pharmacodynamic indicator of target inhibition in patients. Initial phase I studies with PI3K pathway inhibitors have shown some evidence of insulin resistance, as will be described further below, although this has not been a dose- limiting effect. Although p110 and p110 appear to serve distinct functions in insulin signaling, research indicate that p110 mediates glucose homeostasis primarily [73].
Activation of PI3K Signaling in Cancer
In human malignancies, PI3K signaling is triggered in a number of distinct ways. Direct mutational activation or gene amplification of important PI3K pathway genes such PIK3CA and AKT1, as well as PTEN loss, are common causes of increased PI3K signaling. various PI3K signaling pathway components that have undergone genetic changes. Additionally, genetic mutation, upstream RTK amplification, and perhaps Ras that has been activated by mutation can all activate PI3K. The most efficient method of therapy to inhibit the system may be suggested by the mechanism of PI3K activation in a particular malignancy [74, 75, 76, 77, 78, 79, 80].
Somatic Alterations of PI3K Pathway Components in Cancer
Inactivation of the PTEN tumor suppressor gene is the most frequent genetic modification of the PI3K signaling pathway discovered in human cancer. PIP3 builds up as a result of PTEN inactivation because it loses the ability to function as a lipid phosphatase [81]. Protein truncation results from the bulk of somatic mutations in PTEN. Missense mutations, which normally abolish PIP3 phosphatase activity, are also frequently observed, nevertheless [82]. While most PTEN mutations are sporadic, Cowden disease and other hereditary neoplastic diseases have germline PTEN alterations [83]. Human tumors also show homozygous and hemizygous PTEN deletions [84]. Another way of PTEN inactivation involves transcriptional repression and epigenetic silencing, generally by promoter hypermethylation [85]. PTEN loss can have both genetic and epigenetic sources, making it difficult to accurately diagnose a cancer’s PTEN status and possibly requiring proper assessments of protein expression.
Expression of these PIK3CA mutants leads to increased oncogenic potential in vitro and in vivo [86]. They cause constitutive signaling along the PI3K pathway in the absence of growth factors and therefore seem to obviate the usual obligate interactions with tyrosine phosphorylated RTKs and/or adapters. Thus, it is intriguing that some studies have suggested that the presence of these mutations confers resistance to therapies targeting RTKs [87, 88, 89]. When fibroblasts and mammary epithelial cells express mutant PIK3CA, this leads to transformation, growth factor-independent proliferation, and apoptosis resistance [90]. In addition, kinase-domain mutant p110 H1047R transgenic mice with lung-specific induction produce lung adenocarcinomas [91]. Along with these activating mutations, PIK3CA amplification is another frequent finding in ovarian cancer and other cancers; however, it is unclear how amplification impacts PI3K activation.
PI3K Activation by Receptor Tyrosine Kinases and Ras
PI3K is frequently activated in normal epithelial cells after RTK signaling. These RTKs are frequently altered, amplified, or overexpressed in malignancies, which results in abnormal PI3K activation. When RTK-targeting therapies are successful, PI3K signaling is inevitably lost. For instance, PI3K is activated by the human epidermal growth factor receptor 2 (HER2) in breast malignancies with HER2 amplification and the epithelial growth factor receptor (EGFR) in lung tumors having somatic activating mutations in EGFR. In certain tumors, kinase-dead ErbB3 is phosphorylated by EGFR or HER2, which immediately binds and activates PI3K. As a result, when these malignancies are effectively treated with EGFR- and HER2-focused therapies, respectively, PI3K signaling is turned off, and the cells die. Similar to this, glioblastomas typically display PI3K activation, either as a result of the co-activation of several activated RTKs, such as the constitutively active EGFR vIII mutant, or as a result of the loss of PTEN and simultaneous activation of RTKs [92].
Potential Roles for p110, p110, and p110 in Transformation
While oncogenic mutations in p110, p110, or the elegance IB catalytic isoform p110 have not been confirmed, activating mutations in PIK3CA are routinely found in human malignancies. Despite the fact that rare somatic single- residue alterations in p110 and p110 were found (www. sanger.ac.uk/perl/genetics/CGP/cosmic), it is unknown what those substitutions do. Despite the lack of evidence for activating mutations in the various p110 catalytic isoforms, recent research has shown that p110, p110, and p110 have oncogenic potential. Contrary to p110, it’s interesting to note that overexpression of wild-type p110, p110, or p110 transforms cells in culture [93].
Although the and isoforms are typically only expressed in leukocytes, extended p110 (along with p110) and glioblastomas have also been found to express these isoforms. While p110 is increased through the Bcr-Abl oncogene in persistent myelogenous leukemia, p110 appears to provide the crucial PI3K activity in acute myelogenous leukemia [94].
Pi3k Pathway Inhibitors Entering The Clinic: Preclinical And Early Clinical Data
Numerous potential medications that target the PI3K signaling cascade have been developed. AKT inhibitors, dual PI3K-mTOR inhibitors, PI3K inhibitors (that do not inhibit mTOR), and mTOR catalytic site inhibitors are the four different groups of PI3K pathway inhibitors that we shall discuss [95].
Dual PI3K-mTOR Inhibitors
Because they all fall under the phosphatidylinositol kinase-related kinase family of kinases, the p110 subunits and mTOR have catalytic domain names that are structurally identical. There are numerous pharmacologic inhibitors in development that block both the p110 catalytic subunit and mTOR. Dual PI3K-mTOR inhibitors are what these are known as Goel A, et al. [96]. Dual PI3KmTOR inhibitors may have the advantage of inhibiting both mTORC1 and mTORC2 when compared to other types of PI3K pathway inhibitors. Therefore, they should be able to totally shut off this pathway and overcome the feedback inhibition that is typically present with mTORC1 inhibitors (i.e., rapamycin analogs), which can limit their effectiveness. Dual PI3K-mTOR inhibitors may successfully inhibit all p110 isoforms and mTOR, but it is still unknown whether their usage will require sacrificing partial suppression of one or more potential target genes [97].
PI3K Inhibitors
Pan-PI3K inhibitors and isoform-specific inhibitors are two categories of PI3K inhibitors. All class IA PI3K within the cancer are the target of pan-PI3K inhibitors. These are made up of wortmannin prodrugs like the self-activating viridans, which are modified by dextran linker moieties to improve permeability and lengthen serum half-life, or wortmannin derivatives like PX-886 [95, 96, 97].
mTOR Catalytic Site Inhibitors
In mammalian cells, rapamycin and FKBP12 combine to create a complex that directly binds to the mTORC1 FKBP12- rapamycin-binding domain, but not the mTORC2 domain [98]. On the other hand, ATP-competitive mTOR inhibitors work to block the activity of both mTORC1 and mTORC2 by inhibiting the kinase domain of mTOR. Theoretically, inhibiting mTORC2 would have the advantage of preventing AKT activation. Because it would prevent AKT activation, an ATP-competitive mTOR inhibitor might be more effective than rapamycin at preventing the activation of PI3K that frequently follows mTORC1 inhibition [99].
Potential Clinical Uses for PI3K Pathway Inhibitors
HER2-amplified breast cancers, cancers with PIK3CA mutations, and cancers with PTEN deficiency are three types of genetically characterized tumors that PI3K pathway inhibitors have been found to have considerable single-agent action in so far. According to research available so far, PI3K pathway inhibitors may not be as effective against tumors with KRAS mutations. Since many colon cancers include both KRAS and PIK3CA mutations, it appears likely that the presence of KRAS mutations may limit the effectiveness of single-agent PI3K pathway inhibitors in these malignancies [100].
Cellular Functions of the PI3K-Akt Pathway and its Dysregulation in Human Cancer Figure 13
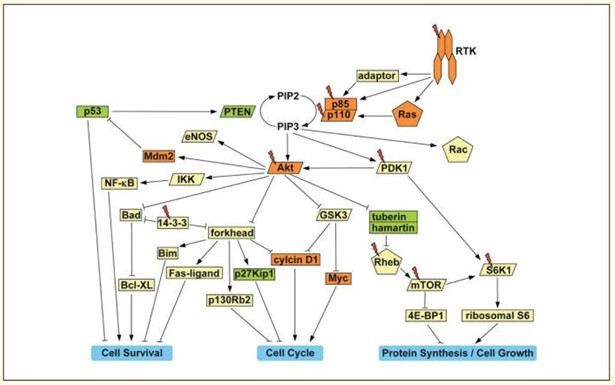
The class-Ia PI3K, a heterodimer made up of the p85 regulatory and p110 catalytic subunits, is activated by growth factor RTKs. The Src-homology 2 (SH2) domains of p85 connect to certain phospho-tyrosine residues on the active receptor or on related adaptor proteins, attracting the enzyme to the membrane. Ras, a small GTPase, can directly bind to p110 and recruit and activate PI3K. Phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) at the 3′ position on its inositol ring by PI3K results in the conversion of PIP2 to PIP3 at the membrane. PIP3 then binds to the pleckstrin homology (PH) domains of many downstream enzymes, most notably the serine-threonine kinases Akt and PDK1. PDK1 phosphorylates threonine 308 in the activation loop of Akt at the membrane, partially activating it. The C terminus of Akt is further phosphorylated at serine 473 to fully activate it [101, 102, 103, 104].
Therapeutic Approaches of Targeting the PI3K-Akt Pathway in Cancer
Potentially many reasons, the PI3K-Akt pathway’s constituents provide potentially promising targets for therapeutic intervention. First, this route inhibits a number of “tumor suppressor-like” proteins that impair cell survival, proliferation, and growth, including the FOXO transcription factors, Bad, GSK3, and the tuberin/hamartin complex. Targeting PI3K directly would be the most effective technique to block the PI3K-Akt pathway. Both wortmannin and the PI3K inhibitors LY294002 and p110’s catalytic site have been widely employed as research tools. It has been demonstrated that LY294002 inhibits tumor growth in vivo in a mouse xenograft model of ovarian cancers. It is not unexpected that neither chemical distinguishes between the different isoforms of PI3K Kinase because inhibitors that target either Akt or PDK1 would also be effective at blocking this pathway because the PI3K catalytic domain is largely conserved among PI3K family members. First-generation inhibitors lack selectivity due to the significant homology of the catalytic domains of AGC family kinases (which include Akt and PDK1). The creation of more focused inhibitors could benefit from the high-resolution crystal structures of the Akt and PDK1 kinase domains [105, 106, 107, 108, 109]. Last but not least, cancers of various origins that develop from hyperactivation of the PI3K-Akt pathway are likely to respond differently to the inhibition of various downstream targets. The efficiency of focusing on a particular section of this pathway will rely, for instance, on which Akt substrates are phosphorylated inside the individual tumor and may also be influenced by the tumor cells’ level of differentiation. Based on this understanding, the therapeutic index of various PI3K-Akt pathway inhibitors could be further improved [110].
References
-
Kushi LH, Doyle C, McCullough M, Rock CL, Demark‐ Wahnefried W, et al. (2012) American Cancer Society Guidelines on nutrition and physical activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA: a cancer journal for clinicians 62(1): 30-67.
-
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267(5203): 1456-1462.
-
Cummings J, Ward TH, Ranson M, Dive C (2004) Apoptosis pathway-targeted drugs— from the bench to the clinic. Biochimica Biophysica Acta (BBA) Reviews on Cancer 1705(1): 53-66.
-
Aydemir N, Bilaloğlu R (2003) Genotoxicity of two anticancer drugs, gemcitabine and topotecan, in mouse bone marrow in vivo. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 537(1): 43- 51.
-
Singh M, Singh SK (2019) Benzothiazoles: how relevant in cancer drug design strategy? Anti-Cancer Agents in Medicinal Chemistry 14(1): 127-146.
-
Keum N, Giovannucci E (2019) Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nature reviews Gastroenterology & hepatology 16(12): 713-732.
-
Murphy N, Moreno V, Hughes DJ, Vodicka L, Vodicka P, et al. (2019) Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Molecular Aspects of Medicine 69: 2-9.
-
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 71(3): 209-249.
-
Arbyn M, Weiderpass E, Bruni L, Sanjosé SD, Saraiya M, et al. (2020) Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. The Lancet Global Healthm 8(2): e191-203.
-
Bray F, Colombet M, Mery L, Piñeros M, Znaor A, et al. (2017) Cancer incidence in Uganda (2008–2012). Cancer incidence in five continents, pp: 11.
-
Vickers NJ (2017) Animal communication: when i’m calling you, will you answer too. Current biology 27(14): R713-R715.
-
Porta C, Paglino C, Mosca A (2014) Targeting PI3K/Akt/ mTOR signaling in cancer. Frontiers in Oncology 4: 64.
-
Sadeghi N, Gerber DE (2012) Targeting the PI3K pathway for cancer therapy. Future medicinal chemistry 4(9): 1153-1169.
-
Carracedo A, Pandolfi PP (2008) The PTEN–PI3K pathway: of feedbacks and cross- talks. Oncogene 27(41): 5527-5541.
-
Cornejo MG, Mabialah V, Sykes SM, Khandan T, Celso CL, et al. (2011) Crosstalk between NOTCH and AKT sign6aling during murine megakaryocyte lineage specification. Blood the Journal of the American Society of Hematology 118(5): 1264-1273.
-
Moreno CS (2010) The Sex-determining region Y-box 4 and homeobox C6 transcriptional networks in prostate cancer progression: crosstalk with the Wnt, Notch, and PI3K pathways. The American journal of pathology 176(2): 518-527.
-
Li T, Wang G (2014) Computer-aided targeting of the PI3K/Akt/mTOR pathway: toxicity reduction and therapeutic opportunities. International journal of molecular sciences 15(10): 18856-18891.
-
Harashima N, Inao T, Imamura R, Okano S, Suda T, et al. (2012) Roles of the PI3K/Akt pathway and autophagy in TLR3 signaling-induced apoptosis and growth arrest of human prostate cancer cells. Cancer immunology immunotherapy 61(5): 667-676.
-
Axanova LS, Chen YQ, McCoy T, Sui G, Cramer SD, et al. (2010) 1, 25‐dihydroxyvitamin D3 and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. The Prostate 70(15): 1658-1671.
-
Wegiel B, Bjartell A, Culig Z, Persson JL (2008) Interleukin‐6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. International journal of cancer 122(7): 1521-1529.
-
Martelli L, Collini P, Meazza C, Bianchi A, Salvini F, et al. (2008) Angiomatoid fibrous histiocytoma in an HIV- positive child. Journal of Pediatric Hematology Oncology 30(3): 242-244.
-
Burris III HA (2013) Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol 71(4): 829-842.
-
Huang J, Zhang L, Chen J, Wan D, Zhou L, et al. (2021) The landscape of immune cells indicates prognosis and applicability of checkpoint therapy in hepatocellular carcinoma. Frontiers in oncology, pp: 3886.
-
Sever R, Brugge JS (2014) Signaling in cancer. Cold Spring Harb Perspect Med 5(4): a006098.
-
Bartholomeusz C, Gonzalez-Angulo AM (2012) Targeting the PI3K signaling pathway in cancer therapy. Expert opinion on therapeutic targets 16(1): 121-130.
-
Yin Y, Shen WH (2008) PTEN: a new guardian of the genome. Oncogene 27(41): 5443-5453.
-
Jian M, Yunjia Z, Zhiying D, Yanduo J, Guocheng J, et al. (2019) Interleukin 7 receptor activates PI3K/Akt/mTOR signaling pathway via downregulation of Beclin‐1 in lung cancer. Molecular Carcinogenesis 58(3): 358-365.
-
Brown VI, Hulitt J, Fish J, Sheen C, Bruno M, et al. (2007) Thymic stromal-derived lymphopoietin induces proliferation of pre- B leukemia and antagonizes mTOR inhibitors, suggesting a role for interleukin- 7Rα signaling. Cancer research 67(20): 9963-9970.
-
Silva A, Girio A, Cebola I, Santos CI, Antunes F, et al. (2011) Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 25(6): 960-9607.
-
Allgäuer A, Schreiner E, Ferrazzi F, Ekici AB, Gerbitz A, et al. (2015) IL-7 abrogates the immunosuppressive function of human double-negative T cells by activating Akt/mTOR signaling. The Journal of Immunology 195(7): 3139-3148.
-
Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, et al. (2004) Activation of PI3K is indispensable for interleukin 7–mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. The Journal of experimental medicine 200(5): 659- 669.
-
Zheng Y, Tan K, Huang H (2019) Retracted: Long noncoding RNA HAGLROS regulates apoptosis and autophagy in colorectal cancer cells via sponging miR‐100 to target ATG5 expression. Cytoskeleton 120(3): 3922-3933.
-
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, et al. (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The lancet 380(9859): 2095-2128.
-
Torrealba N, Rodriguez-Berriguete G, Fraile B, Olmedilla G, Martínez-Onsurbe P, et al. (2018) PI3K pathway and Bcl-2 family. Clinicopathological features in prostate cancer. The Aging Male 21(3): 211-222.
-
Katzung BG (2017) Basic and clinical pharmacology. Access Medicine, McGraw Hill, USA, pp: 1265.
-
Shinde A, Hardy SD, Kim D, Akhand SS, Jolly MK, et al. (2019) Spleen Tyrosine Kinase–Mediated Autophagy Is Required for Epithelial–Mesenchymal Plasticity and Metastasis in Breast CancerSYK Activity Facilitates Mesenchymal–Epithelial Transition. Cancer research 79(8): 1831-1843.
-
Shinde A, Libring S, Alpsoy A, Abdullah A, Schaber JA, et al. (2018) Autocrine Fibronectin Inhibits Breast Cancer Metastasis Autocrine Fibronectin Limits Metastasis. Molecular Cancer Research 16(10): 1579-1589.
-
Hardy SD, Shinde A, Wang WH, Wendt MK, Geahlen RL, et al. (2017) Regulation of epithelial-mesenchymal transition and metastasis by TGF-β, P-bodies, and autophagy. Oncotarget 8(61): 103302-103314.
-
Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, et al. (2013) First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer discovery 3(4): 406-417.
-
Zhu J, Sun Y, Lu Y, Jiang X, Ma B, et al. (2018) Glaucocalyxin A exerts anticancer effect on osteosarcoma by inhibiting GLI1 nuclear translocation via regulating PI3K/Akt pathway. Cell death & disease 9(6): 1-6.
-
Jin S, Pang RP, Shen JN, Huang G, Wang J, et al. (2007) Grifolin induces apoptosis via inhibition of PI3K/ AKT signalling pathway in human osteosarcoma cells. Apoptosis 12(7): 1317-1326.
-
Lytton SD, Antiga E, Pfeiffer S, Matthias T, Szaflarska- Poplawska A, et al. (2013) Neo-epitope tissue transglutaminase autoantibodies as a biomarker of the gluten sensitive skin disease—dermatitis herpetiformis. Clinica Chimica Acta 415: 346-349.
-
Hu K, Dai HB, Qiu ZL (2016) mTOR signaling in osteosarcoma: Oncogenesis and therapeutic aspects. Oncology reports 36(3): 1219-1225.
-
Curigliano G, Shah RR (2019) Safety and tolerability of phosphatidylinositol-3-kinase (PI3K) inhibitors in oncology. Drug safety 42(2): 247-262.
-
Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, et al. (2003) Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 17(5): 995-997.
-
Chen W, Drakos E, Grammatikakis I, Schlette EJ, Li J, et al. (2010) mTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Molecular cancer 9(1): 1-7.
-
Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, et al. (2009) Functional proteomic profiling of AML predicts response and survival. Blood The Journal of the American Society of Hematology 113(1): 154-164.
-
Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, et al. (2008) Abecasis M, Nowill AE, Leslie NR, Cardoso AA, Barata JT PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. The Journal of clinical investigation 118(11): 3762-3774.
-
Niemann CU, Jones J, Wiestner A (2013) Towards targeted therapy of chronic lymphocytic leukemia. Advances in Chronic Lymphocytic Leukemia 792: 259-291.
-
Nyåkern M, Tazzari PL, Finelli C, Bosi C, Follo MY, et al. (2006) Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia 20(2): 230-238.
-
Hyun T, Yam A, Pece S, Xie X, Zhang J, et al. (2000) Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood The Journal of the American Society of Hematology 96(10): 3560-3568.
-
Hu L, Shi Y, Hsu JH, Gera J, Ness BV, et al. (2003) Downstream effectors of oncogenic ras in multiple myeloma cells. Blood The Journal of the American Society of Hematology 101(8): 3126-3135.
-
Gao Y, Yuan CY, Yuan W (2016) Will targeting PI3K/Akt/ mTOR signaling work in hematopoietic malignancies. Stem Cell Investigation 3: 31
-
Blackadar CB (2016) Historical review of the causes of cancer. World journal of clinical oncology 7(1): 54.
-
Saini A, Kumar M, Bhatt S, Saini V, Malik A, et al. (2020) Cancer causes and treatments. International Journal of Pharmaceutical Sciences and Research 11: 3109-3122.
-
Leitch A (1923) A British Medical Association Lecture on the experimental inquiry into the causes of cancer. British Medical Journal 2(3262): 1-7.
-
Jia S, Roberts TM, Zhao JJ (2009) Should individual PI3 kinase isoforms be targeted in cancer Current. Opinion in cell biology 21(2): 199-208.
-
Kaplan DR, Whitman M, Schaffhausen B, Pallas DC, White M, et al. (1987) Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 50(7): 1021-1029.
-
Sugimoto Y, Whitman M, Cantley LC, Erikson RL (1984) Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proceedings of the National Academy of Sciences 81(7): 2117-2121.
-
Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM, et al. (1985) Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315(6016): 239-242.
-
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics 7(8): 606- 619.
-
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, et al. (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304(5670): 554.
-
Yuan TL, Cantley L (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27(41): 5497- 5510.
-
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, et al. (2001) Cellular function of phosphoinositide 3-kinases: implications for development, immunity, homeostasis, and cancer. Annual review of cell and developmental biology 17(1): 615-675.
-
Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, et al. (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer research 65(11): 4562- 4567.
-
Mizoguchi M, Nutt CL, Mohapatra G, Louis DN (2004) Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 14(4): 372- 377.
-
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, et al. (2001) The phosphatidylinositol 3-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 61(20): 7426-7429.
-
Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell cycle 3(10): 1221-1224.
-
Kang S, Denley A, Vanhaesebroeck B, Vogt PK (2006) Oncogenic transformation induced by the p110β,-γ, and-δ isoforms of class I phosphoinositide 3-kinase. Proceedings of the National Academy of Sciences 103(5): 1289-1294.
-
Skolnik EY, Margolis B, Mohammadi M, Lowenstein E, Fischer R, et al. (1991) Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell 65(1): 83-90.
-
Zhao L, Vogt PK (2008) Class I PI3K in oncogenic cellular transformation. Oncogene 27(41): 5486-5496.
-
Carpenter CL, Auger KR, Chanudhuri M, Yoakim M, Schaffhausen B, et al. (1993) Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. Journal of biological chemistry 268(13): 9478-9483.
-
Shaw RJ, Cantley LC (2006) Ras, PI (3) K and mTOR signalling controls tumour cell growth. Nature 441(7092): 424-430.
-
Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, et al. (2004) Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. The EMBO journal 23(20): 3918-3928.
-
Currie RA, Walker KS, Gray A, Deak M, Casamayor A, et al. (1999) Role of phosphatidylinositol 3, 4, 5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochemical Journal 337(3): 575-583.
-
Majumder PK, Sellers WR (2005) Akt-regulated pathways in prostate cancer. Oncogene 24(50): 7465-7474.
-
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296(5573): 1655-1657.
-
Duronio V (2008) The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochemical Journal 415(3): 333-344.
-
Ma WW, Adjei AA (2009) Novel agents on the horizon for cancer therapy. CA: a cancer journal for clinicians 59(2): 111-137.
-
Meric-Bernstam F, Gonzalez-Angulo AM (2009) Targeting the mTOR signaling network for cancer therapy. Journal of Clinical Oncology 27(13): 2278-2287.
-
Luo J, Sobkiw CL, Hirshman MF, Logsdon MN, Li TQ, et al. (2006) Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell metabolism 3(5): 355-366.
-
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, et al. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292(5522): 1728-1731.
-
Taniguchi CM, Tran TT, Kondo T, Luo J, Ueki K, et al. (2006) Phosphoinositide 3-kinase regulatory subunit p85α suppresses insulin action via positive regulation of PTEN. Proceedings of the National Academy of Sciences 103(32): 12093-12097.
-
Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, et al. (2002) Increased insulin sensitivity in mice lacking p85β subunit of phosphoinositide 3-kinase. Proceedings of the National Academy of Sciences 99(1): 419-424.
-
Jia S, Liu Z, Zhang S, Liu P, Zhang L, et al. (2008) Essential roles of PI (3) K–p110β in cell growth, metabolism and tumorigenesis. Nature 454(7205): 776-779.
-
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, et al. (2006) A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 125(4): 733-747.
-
Bader AG, Kang S, Vogt PK (2006) Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proceedings of the National Academy of Sciences 103(5): 1475-1479.
-
Li J, Yen C, Liaw D, Podsypanina K, Bose S, et al. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308): 1943-1947.
-
Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, et al. (1999) PIK3CA is implicated as an oncogene in ovarian cancer. Nature genetics 21(1): 99-102.
-
Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, et al. (2009) The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. British journal of cancer 101(1): 145-148.
-
Obata K, Morland SJ, Watson RH, Hitchcock A, Chenevix- Trench G, et al. (1998) Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer research 58(10): 2095-2097.
-
Han SY, Kato H, Kato S, Suzuki T, Shibata H, et al. (2000) Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer research 60(12): 3147-3151.
-
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, et al. (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature genetics 16(1): 64-67.
-
Agell L, Hernández S, Salido M, Muga SD, Juanpere N, et al. (2011) PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Modern Pathology 24(3): 443-452.
-
García JM, Silva J, Peña C, Garcia V, Rodríguez R, et al. (2004) Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes and Cancer 41(2): 117-124.
-
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, et al. (2004) Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability- high sporadic colorectal cancers. Cancer research 64(9): 3014-3021.
-
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, et al. (2007) A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer cell 12(4): 395-402.
-
Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borrás AM, et al. (2006) Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. The Journal of clinical investigation 116(10): 2695-2706.
-
Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, et al. (2005) Breast cancer–associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer research 65(23): 1000-10992.
-
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, et al. (2005) The oncogenic properties of mutant p110α and p110β phosphatidylinositol 3-kinases in human mammary epithelial cells. Proceedings of the National Academy of Sciences 102(51): 18443-18448.
-
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, et al. (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature medicine 14(12): 1351-1356.
-
Engelman JA, Settleman J (2008) Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Current opinion in genetics & development 18(1): 73-79.
-
Eichhorn PJA, Gili M, Scaltriti M, Serra V, Guzman M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP- BEZ235. Cancer research 68(22): 9221-9230.
-
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas III CF, et al. (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences 100(15): 8933-8938.
-
Moasser MM, Basso A, Averbuch SD, Rosen N (2001) The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer research 61(19): 7184-7188.
-
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, et al. (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. New England Journal of Medicine 353(19): 2012-2024.
-
Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, et al. (2008) p53 and Pten control neural and glioma stem/ progenitor cell renewal and differentiation. Nature 455(7216): 1129-1133.
-
Hickey FB, Cotter TG (2006) BCR-ABL regulates phosphatidylinositol 3-kinase- p110γ transcription and activation and is required for proliferation and drug resistance. Journal of Biological Chemistry 281(5): 2441-2450.
-
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, et al. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research 66(3): 1500-1508.
-
Blois J, Yuan H, Smith A, Pacold ME, Weissleder R, et al. (2008) Slow self-activation enhances the potency of viridin prodrugs. Journal of medicinal chemistry 51(15): 4699-4707.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review