Bioequivalence Study of Salbutamol 400 mg Tablet in Healthy Indonesian Volunteers by Liquid Chromatography Tandem with Mass Spectrometry
This study objective was to determine the bioequivalence of Salbutamol 4 mg Tablet manufactured by PT Kimia Farma Tbk compared to Ventolin® 2x2 mg Tablet manufactured by Glaxo Wellcome Indonesia, in healthy Indonesian volunteer. The pharmacokinetic parameters calculated in this study are AUC0-24, AUC0-inf, Cmax, Tmax, and T1/2. The study was conducted in a randomized, single-dose, open-label, two-way crossover design (2 treatments, 2 periods, and 2 sequences) under fasting state with 7 (seven) days washed-out period. The number of subjects who participated in the study were 24 volunteers ((12 males and 12 females). The participants were provided with an overview of the study and signed the informed consent form. The study participants underwent a minimum 8-hour fasting period prior to receiving both the test drug and the reference drug, and blood samples were collected at 14 specified time points: 0 hours (before drug administration), minutes-20, minutes-40, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 hours after drug administration. Plasma concentrations of the drug were determined by LCMS/MS method. The acceptance criteria for the bioequivalence test are 80.00 - 125.00% for AUC0-t and Cmax with 90% Confidence Interval (CI) with α = 5.00%. The mean SD value of AUC0-24, AUC0-inf, Cmax, Tmax, and T1/2 respectively for test drug is 84.49 ±18.99 ng.mL-1.hour; 89.66 ±21.29 ng.mL-1.hour; 13.85 ±4.25 ng/mL; 2.00 ±0.71 hour and 5.08 ±1.27 hour. The mean SD values of AUC0-24, AUC0-inf, Cmax, Tmax, and C1/2 respectively for reference drug are 88.81 ±26.50 ng.mL-1.jam; 93.78 ±27.97 ng.mL-1.jam; 13.39 ±5.39 ng/mL; 2.63 ±1.45 hour and 5.03 ±1.16 hour. Meanwhile, the geometric mean ratio of test drug to reference drug (90% confidence interval) is 96.35% (90.55- 102.53%) for AUC0-24 and 105.39% (93.62-118.65%) for Cmax. Based on the AUC0-24 and Cmax values, Salbutamol 4 mg Tablet manufactured by PT Kimia Farma Tbk is bioequivalent to Ventolin® 2x2 mg Tablet manufactured by Glaxo Wellcome Indonesia.
Introduction
The chemical name of Salbutamol is 1,3-Benzenedimethanol, alpha1-[[(1,1-dimethylethyl) amino] methyl]-4-hydroxy-sulfate (2:1) (salt). The molecular formula of Salbutamol is (C13H21NO3)2.H2SO4 [1].
Salbutamol is a β2-adrenoceptor stimulant that causes muscle relaxation smooth respiratory tract through activation of cyclic adenyl and increase in intracellular cyclic3′,5′- adenosine monophosphate (cyclic AMP). Salbutamol inhibits myosin phosphorylation causes bronchial muscles and uterine relaxation, peripheral vessels dilate, heart rate increases, and metabolic effects (e.g. decreased plasma potassium levels). β2-adrenoreceptors are the predominant adrenergic receptors in bronchial smooth muscle, while β1- adrenoceptors in heart. The number of β2-adrenoceptors in the human heart consists of 10% to 50% of total β-adrenoceptors. Salbutamol also has certain significant anti-inflammatory properties clinically unknown [2]. Salbutamol is indicated for symptom relief and prevention bronchospasm in bronchial asthma, chronic bronchitis, and emphysema [3].
In oral administration, Salbutamol will be well absorbed through the gastrointestinal tract. Salbutamol is absorbed rapidly after oral administration at a dose of 2-4 mg with the tmax value about 2-3 hours and Cmax 14 ng/mL to 18 ng/mL [4]. Salbutamol does not metabolize in the lungs but in the liver to its metabolite, namely 4’-o-sulfate ester (salbutamol 4’-O-sulfate). Salbutamol can also be metabolized by oxidative deamination by conjugation with glucuronide. Salbutamol is excreted in the urine as free drug or its metabolites. Half- life (t1/2) of Salbutamol elimination is between 2.7-5 hours and the terminal plasma half-life is around 4.6 hours. After oral administration, 58-78% of the dose is excreted in urine within 24 hours, about 60% as metabolites. Only a small portion of Salbutamol is excreted through feces [2].
Salbutamol may cause various side effects, such as tremors, headaches, peripheral vasodilation, tachycardia, and muscle cramps [3]. Additionally, it can lead to a rapid or pounding heartbeat, restlessness in the legs, arms, or hands. Rare side effects encompass pain in other limbs, the back, or lower jaw, chest tightness or heaviness, confusion, dizziness, fainting, and a feeling of intoxication [5].
Salbutamol should not be administered to individuals with hypersensitivity to Salbutamol. Exercise caution when prescribing Salbutamol to patients with cardiovascular conditions, such as ischemic heart disease (coronary artery disease), hypertension, cardiac arrhythmia, tachycardia, or QT interval prolongation. Patients with a history of seizure disorders, hyperthyroidism, diabetes mellitus, and diabetic ketoacidosis should also be treated with care. The use of Salbutamol during pregnancy and breastfeeding is not advisable. Do not exceed the recommended dosage of Salbutamol, as excessive inhalation use has been associated with reported deaths in asthma patients [6].
This study objective was to determine the bioequivalence of Salbutamol 4 mg Tablet manufactured by PT Kimia Farma Tbk compared to Ventolin® 2x2 mg Tablet manufactured by Glaxo Wellcome Indonesia, in healthy Indonesian volunteer.
Study Protocol
The protocol study has received approval from the Ethics Committee of the Faculty of Medicine, University of Indonesia and the Indonesian Food and Drug Regulatory Authority. The protocol describes all project details, including study design, clinical procedures, bioanalysis, pharmacokinetic and statistical data analysis, bioequivalence evaluation, informed consent form, publication of the final report, and complete documentation.
Ethical Considerations
The Ethics Committee at the University of Indonesia’s Medical Faculty, certified by the Forum for Ethical Review Committee in the Asia and Western Pacific Region (FERCAP), has thoroughly assessed the protocol and informed consent. The principal investigator for the study is required to maintain communication with the ethics committee in the event of any alterations to the protocol, encompassing changes in the study’s procedures, benefits, or risks. If there are modifications to the study’s objectives, design, sample size, or other critical aspects that impact the study, protocol amendments will be implemented. These amendments are subject to review and approval by both the Ethics Committee and BPOM.
During the screening phase before the study commenced, every participant received the informed consent. The principal investigator and clinical investigators organized a meeting to elucidate all aspects of the study to the participants. This encompassed the study’s objectives, potential risks and benefits, procedures, the participant’s right to withdraw at any point during the study, and the compensation in the event of any harm resulting from the study.
Study Design
The study was conducted in a randomized, single-dose, open-label, two-way crossover design (2 treatments, 2 periods, and 2 sequences) under fasting state with 7 (seven) days washed-out period [7]. The subjects were randomly assigned to each dosing sequence of the investigational drug products (test and reference drug). The subjects who took part in the study were 24 people (12 male and 12 female).
Inclusion and Exclusion Criteria for Participation in the Study
Based on the following inclusion criteria, the subjects were considered eligible for participation in this study: 1) willing to sign an informed consent; 2) Healthy based on clinical laboratory tests (routine haematology, liver function, kidney function, blood glucose, urinalysis, hepatitis B (HBsAg), hepatitis C (Anti-HCV) and HIV (Anti-HIV), medical history, and physical examination); 3) Male and female subjects (if female, consider the risks for women of childbearing age and perform pregnancy tests); 4) Age between 18-55 years; 5) Normal weight range according to Body Mass Index (BMI) 18-25 kg/m2); 6) Vital signs within the following ranges: systolic blood pressure 110-129 mmHg, diastolic blood pressure 70-84 mmHg, normal pulse rate 60- 90 bpm, oxygen saturation (SpO2) in the normal range of 95- 100%, and normal respiratory rate of 12-20/min [8].
The exclusion criteria for this study including: 1) Smoking more than 10 cigarettes per day; 2) Pregnant or breastfeeding women (Pregnancy tests was performed during screening and prior to the administration of the investigational or comparator drug); 3) History of kidney or liver disease, or history of allergy, hypersensitivity or contraindication to the investigational bioequivalence drug (Salbutamol); 4) Clinically significant haematological abnormalities; 5) Abnormal electrocardiogram (ECG); 6) Difficulty accessing veins in the left or right arm; 7) History of significant ongoing clinically or medically significant chronic or acute illness; 8) History of drug or alcohol abuse within the past 12 months (1 year) prior to screening for this study; 9) Positive serology test results for Hepatitis B (HBsAg), Hepatitis C (anti-HCV), HIV (anti-HIV). 10) Positive rapid antigen test results for SARS-CoV-2 (if the BE study is conducted during a pandemic); 11) Have history or condition that can affect drug kinetics; 12) Use of drugs or dietary supplements no more than 7 days since the start of the study; 13) participated in previous clinical trials no more than 3 months from the start of the study; and 14) Blood donation or blood loss of more than
300 ml within 3 months from the start of the study [8].
Health Screening
Prior to the study, a health screening is carried out to assess the subjects’ health condition, in accordance with the inclusion and exclusion criteria. Within seven days before the first administration of the study drug, subjects undergo a comprehensive medical examination. This examination encompasses a thorough physical assessment, measurement of vital signs (such as blood pressure, pulse rate, and body temperature), and an ECG conducted by the Responsible Physician at Equitrust Lab. Clinical Laboratory handles the testing of routine haematological values, liver and kidney function, blood glucose levels, and urinalysis. Additionally, Equitrust Lab conducts immunology tests for HBsAg, HCV, HIV, and Rapid Test Antigen for Covid-19. During the screening and immunology tests (HBsAg, HCV, and HIV), approximately 10 mL of blood samples are collected from each subject.
Drug Product Administration
This study consists of two periods with 24 hours for each period. At the specified time, the subjects will be asked to come to the Equitrust Lab on a day before drug administration for quarantine. Subjects were asked not to take medication or supplements for at least 1 week before the study began. If the subject is in an urgent situation and are forced to take medication or supplements, the medication or supplement taking and the amount of the dose must be reported to the investigator.
The subjects must fast for 8 hours before taking the investigational drug or reference drug. About 1 hour before taking the medicine, the subject’s condition was checked by a doctor to check their health. Inspection carried out blood pressure, pulse rate, body temperature, oxygen saturation (SpO2) examination, and respiratory rate. The results of the examination are recorded in the form of a Case Report Form (CRF). For female subjects, additional examinations are carried out, such as pregnancy tests. Starting at 07.00 a.m., on the first day of sampling (0 hours), the subject was given the test drug or Comparator drug with 240 mL of water in a sitting position. Subjects were asked to maintain an upright position, either standing or sit for 1 hour after administering the drug.
After 2 hours, water was provided. No food was allowed until 4 hours after drug administration. Standard meals were reserved for 4 hours (lunch), 8 hours (snack), and 12 hours (dinner) after study drug administration. Subjects were remaining in a sitting position until 4 hours period after drug administration. The clinical facility did not permit subjects to exit.
Blood Samples Collection
Blood samples were taken at specific time points to represent the drug absorption, distribution, and elimination phases. Most drugs require 12-18 blood samples, including one sample before dosing (t1), 2-3 samples before Cmax, 4-6 samples around Cmax, and 5-8 samples after Cmax [8]. In the Salbutamol sampling, blood samples were taken for 24 hours. A pre-dose blood sample was taken at 6.30 a.m within 30-minutes prior to drug administration and a 5 mL of blood was collected through venipuncture using a syringe at the following time points: 0 hours (before drug administration), minutes-20, minutes-40, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 hours after drug administration (14 points). Every deviation exceeding the predetermined time is recorded in the Case Report Form (CRF) as a protocol deviation.
The blood samples are collected using a syringe and transferred into CPDA blood collection tubes. Plasma is centrifuged at 3000 rpm for 10 minutes and immediately transferred plasma into clean microtubes. Plasma separation was carried out in preparation room at room temperature. Then, the plasma is stored in the freezer at a maximum temperature of -20ºC until Salbutamol analysis is conducted. The total amount of blood collected from each subject was 155 mL (including 15 mL for initial health screening).
Statistical Analysis
Pharmacokinetic parameters, including Cmax, AUC0-t, AUC0-inf, tmax, and t1/2, were calculated for each subject and each period using the statistical program. Bioequivalence between the test and reference products was determined based on the average ratios of Cmax and AUC0-t with a 90% Confidence Interval (90% CI) of the log or ln-transformed data. The log or ln-transformed values of Cmax and AUC0-t for the two products are analysed using a two-way Analysis of Variance (ANOVA) and R program. The compared factors were the drug products (Test and Reference), drug administration period (I and II), subject, and sequence (TR and RT). The mean differences in Cmax and AUC0-t between the test and reference products were considered bioequivalent if the ratio of the geometric mean (AUC)T/(AUC)R= 1.00 with 90% CI= 80.00-125.00% (α: 0.05) and (Cmax)T/(Cmax)R= 1.00 with 90% CI= 80.00-125.00% (α: 0.05). The study had a power of 80% with a significance level (alpha) of 5% (two- tailed) [9].
The 90% Confidence Interval (90% CI) is calculated using the following equation: (90% CI) diff = Difference ± t0.1 (n-2) × SE diff Difference : the mean ln T - the mean ln R n : the number of subjects α : 0.05 SE diff = [(½ MSresidual × (1/nTR + 1/nRT))½] The 90% CI for the ratio is calculated as follows: (90% CI) ratio = antilog (90% CI) diff × 100%
Assay Methodology and Validation
Prior to the analysis of salbutamol assay in the plasma sample, bioanalytical method validation was evaluated for selectivity; carry-over effect; calibration curve and Lower Limit of Quantification (LLOQ); precision and accuracy; matrix effect; dilution integrity; and stabilities (i.e., freeze- thawed stability, short-term stability at room temperature, post preparative stability / autosampler batch integrity, stock solution and internal standard stability, and long-term stability) [10]. The anticoagulant used during the validation and bioanalytical phase was CPDA (Citrate Phosphate Dextrose Adenine).
The analysis of Salbutamol assay in plasma was carried out by a fully validated method with LLOQ 0.2 ng/mL. Plasma containing salbutamol was added with an Aripiprazole as internal standard. Protein precipitation extraction was carried out using acetonitrile. The clear supernatant solution was combined with the mobile phase, and then injected into the LCMS/MS chromatography system [11, 12].
Data Quality Assurance
During the bioanalytical phase of the plasma samples, the analysis was monitored according to the quality control process, which included system suitability test, linearity of calibration curve, and quality control samples (Low QC, Medium QC, and High QC) referred to European Medicinal Agency (EMA) guideline 2011 [10].
The calibration curve will be conducted with eight concentrations at each batch. The acceptance criteria are %LLOQ deviation + 20.00% and %other concentration deviation + 15.00% with a minimum of 75% of the calibration curve with at least 6 concentration levels must meet these criteria. QC Samples will be conducted with three concentrations at each batch. The acceptance criteria are %deviation value and RSD of QC samples must be <15% with a minimum of 67% of the total QC samples and a minimum of 50% at each concentration level must meet these criteria.
The investigator has conducted the study in accordance with the protocol requirements, Good Clinical Practice (GCP), and Good Laboratory Practice (GLP) issuance by the National Agency of Drug and Food Control. The Quality Assurance (QA) department is responsible for conducting regular audits of the BA/BE study to ensure the integrity of the study. The QA department maintains written records and audits findings, and other document related to the study. Any study issues that arise will be evaluated or audited to ensure the integrity of the study data that will be communicated to the investigator. The evaluation results can then be utilized to address any encountered problems. A representative from the sponsor’s QA department will oversee that there are no deviations from the approved protocol or standard operating procedures applied in the study.
Result and Discussion
The total number of subjects who participated the study was 24 subjects (12 males and 12 females). There are no subject dropped out in the study. The demographic data of the subject are tabulated in Table 1.
| MIN | MAX | |
|---|---|---|
| Age (Year) | 19 | 53 |
| Body Weight (kg) | 41 | 66 |
| Body Height (m) | 147 | 173 |
| BMI (kg/m2) | 18.22 | 25 |
Table 1: Demographic Data of 24 subjects.
Several subjects reported an adverse event, namely chest palpitations after administration of the test drug or comparison drug. This is due to salbutamol works on the body’s beta-adrenergic receptors. Salbutamol does not only specifically work on beta 2 receptors (respiratory smooth muscles), but also works on beta 1 receptors (muscles heart). Then the heart muscle will receive stimulation from the drug so that it increases heart rate (chest pounding). However, not all subjects experience this (it depends sensitivity of each subject) and this incident is not disturbing and does not significant effect on the subject’s health. All of these adverse events were recorded in the CRF and the deviation during the study is reported in protocol deviation point at full study report.
Bioanalytical Result
Total plasma sample from 24 subjects is 672 samples (24 individuals x 14-time points x 2 periods). The samples were stored in a freezer at a maximum temperature of -20ºC until analysis. The analysis is conducted on November 15th – 27th, 2021. Sample analysis was performed using a validated analytical method which complied to the Guideline on Bioanalytical Method Validation, EMEA 2011. The bioanalysis result of system suitability test, linearity of calibration curve, and quality control a sample (Low QC, Medium QC, and High QC) was met the requirements.
Pharmacokinetic Analysis
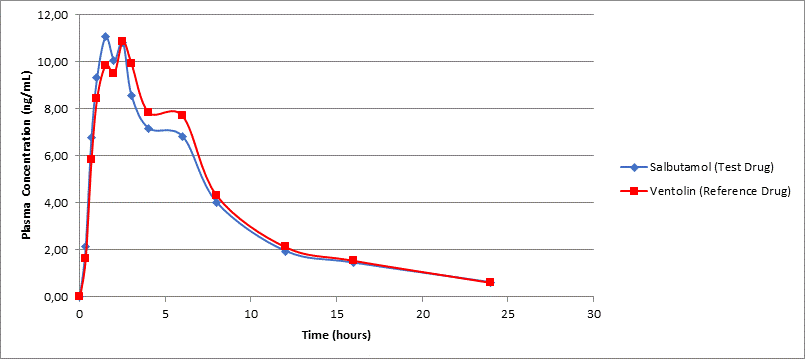
The pharmacokinetic parameters (AUC0-t, AUC0-inf, Cmax, t½, and tmax) of the test drug and comparator drug were calculated and compared to assessed bioequivalence. The calculated 90% CI with a = 5.00% for geometric mean of individual and the ratios of AUC0-inf and AUC0-t as well as Cmax for the test drug Salbutamol and reference drug Ventolin were all within 80.00 - 125.00% interval. This was in conformity with the standard guideline for bioequivalence study [8]. The Summary of pharmacokinetic parameters of the study shown in Table 2. Meanwhile, the main statistical calculations for AUC0-t and Cmax parameter of study Salbutamol was obtained from 24 subjects after oral administration of the test drug and reference drug shown in Table 3. The means of plasma concentration vs. time profiles after a single dose of oral administration of investigational products are shown in Figure 1.
| Parameter | Test Drug | Comparator drug | ||
|---|---|---|---|---|
| Arithmetic | Standard | Arithmetic | Standard | |
| Mean | Deviation | Mean | Deviation | |
| AUC0-24 (ng.h.mL-1) | 84.49 | 18.99 | 88.81 | 26.5 |
| AUC0-inf (ng.h.mL-1) | 89.66 | 21.29 | 93.78 | 27.97 |
| C (ng.mL-1) max | 13.85 | 4.25 | 13.39 | 5.39 |
| t (h) ½ | 5.08 | 1.27 | 5.03 | 1.16 |
| t (h)* max | 2.00 (1.00 – 3.00) | 2.63 (1.00 – 6.00) | ||
| Parameter | % Ratio of Geometric Means (T/R) | 90% Confidence Interval (T/R) | % CV | |
| Lower Limit | Upper Limit | |||
| AUC 0-24 | 96.35 | 90.55 | 102.53 | 12.6 |
| C max | 105.39 | 93.62 | 118.65 | 24.3 |
Table 2: The Summary of pharmacokinetic parameters. *mean (range)
Conclusion
This study objective was to determine the bioequivalence of Salbutamol 4 mg Tablet manufactured by PT Kimia Farma Tbk compared to Ventolin® 2x2 mg Tablet manufactured by Glaxo Wellcome Indonesia, in healthy Indonesian volunteer. The study was conducted in a randomized, single-dose, open-label, two-way crossover design (2 treatments, 2 periods, and 2 sequences) under fasting state with 7 (seven) days washed-out period. The number of subjects who participated in the study were 24 adult male and female volunteers. The subjects received an explanation of the study and signed informed consent. The primary parameters to assess the bioequivalence between the test and reference drug of Salbutamol are AUC0-24 and Cmax. Based on the bioequivalence criteria of a 90% confidence interval, the ratio of the geometric mean values (AUC)T/(AUC)R=1.00 with 90% CI=(80-125)% (α: 0.05) and (Cmax)T/(Cmax)R=1.00 with 90% CI=(80-125)% (α: 0.05) were used.
The geometric mean ratio of test drug to reference drug for Salbutamol (90% confidence interval) were 96.35% (90.55- 102.53%) for AUC0-24 and 105.39% (93.62- 118.65%) for Cmax. Based on the AUC0-24 and Cmax values, Salbutamol 4 mg Tablet manufactured by PT Kimia Farma Tbk is bioequivalent to Ventolin® 2x2 mg Tablet manufactured by Glaxo Wellcome Indonesia. In this study, the intra-subject coefficient of variation (%CV) obtained from ANOVA was
12.58% for AUC0-24 and 24.25% for Cmax.
References
-
United States Pharmacopeia (USP) 29 NF-24 (2023) Albuterol Sulfate. Monograph, The United States Pharmacopeia Convention, pp: 63.
-
Drugbank (2020) Salbutamol.
-
MIMS (2020) Drug Reference, Brief Information on Medicinal Products in Bahasa Indonesia. 21st(Edn.), Bhuana Ilmu Populer, Jakarta, Indonesia, pp: 79.
-
Prescribers Digital Reference (2020) Albuterol.
-
Durbin K (2022) Albuterol Inhalation. Drugs.com.
-
PDR ConnectiveRx (2020) Albuterol Tablets.
-
Chik Z, Basu RC, Pendek R, Lee TC, Mohamed Z, et al. (2009) Comparative bioavailability study of two salbutamol tablets in healthy adult volunteers. Int J Clin Pharmacol Ther 47(6): 413-418.
-
FDRA (2019) Bioequivalence Study Guidelines. Indonesian Food & Drug Regulatory Authority, Jakarta, Indonesia.
-
FDRA (2015) Guidelines for Approval of Clinical Trials. Indonesian Food & Drug Regulatory Authority, Jakarta, Indonesia.
-
European Medicines Agency (2011) Guideline on Bioanalytical Method Validation.
-
Ren Y, Yang J (2012) Determination of Common Beta- Agonist Residues in Meat Products by UPLC-MS/MS. Waters the Science of What’s Possible, USA, pp: 1-5.
-
Snehalatha B, Shankar DG, Rathod S (2014) Development and Validation of a HPLC/MS/MS Method for the Determination of Albuterol in Human Plasma. Pharmacophore 5(5): 647-656.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review