Case Report of Myasthenia Gravis Complicated by Myasthenic Crisisdiagnosedin A Resource-Low Setting
Introduction: Myasthenia gravis is an extremely rare disorder. It may present uniquely with ocular symptoms or it may present with ocular, bulbar and respiratory symptoms. 15-20% of patients with myasthenia gravis are affected by myasthenia crisis at least once in their life time. Case summary: We present the case of a 26 year old male diagnosed of generalized myasthenia gravis in 2015 after presenting predominantly with ocular symptoms but and non-compliant to treatment who later presented with myasthenic crisis dominated by respiratory and ocular symptoms. He was hospitalized in ICU with respiratory and life support. Conclusion: Myasthenia gravis and myasthenic crisis are relatively rare disorders. Presentation can range from ocular through generalized to respiratory failure. This case highlights the importance of considering neuromuscular disorders in patients presenting with ocular symptoms such as diplopia.
Introduction
Myasthenia gravis (MG) is an autoimmune disorder affecting neuromuscular transmission, characterized by localised or generalized weakness and easy fatigability of skeletal muscles [1]. MG is a rare disorder with a prevalence of 50 – 125 cases per million population [2]. Myasthenia gravis is most frequently associated with antibodies against acetylcholine receptors (AChR) in the post-synaptic motor end plate. It may also be associated with antibodies against muscle-specific tyrosine kinase (MuSK) or rarely may it be seronegative, hence, lacking antibodies to AChR and MuSK [3, 4]. Ocular myasthenia (OM) is a subtype of MG with weakness that is isolated to periocular muscles including extraocular muscles, levatorpalpebrae superior is and orbicularis ocular muscles whereas generalized MG involved the bulbar, limb and respiratory muscles [5]. Although, more than half of all MG present initially with ocular symptoms, only 12-15% continue and maintained only ocular symptoms [6]. Myasthenic crisis (MC) is a complication of MG characterized by worsening muscle weakness, resulting in respiratory failure that requires intubation and mechanical ventilation [1]. 15-20% of myasthenic patients are affected by MC at least once in their life time [1]. The median time to first MC from onset of MG ranges from 8-12 months. However, myasthenic crisis may be the initial presentation of MG in one-fifth of patients [7]. MC is a life-threatening medical emergency requiring early diagnosis and respiratory assistance. Mortality in MC has improved considerably largely due to improvement in respiratory care and intensive care unit management. The widespread use of immunotherapy, such as plasmapheresis and intravenous immunoglobulin has substantially altered the management of MC [8]. The authors described their experience in the management of the case of a 26 year old male diagnosed of generalized myasthenia gravis in 2015 after presenting predominantly with ocular symptoms but non-compliant to treatment who later presented with myasthenic crisis dominated by respiratory and ocular symptoms. He was hospitalized in ICU with respiratory and life support.
Case Presentation
A 26 year old male sub-Sahara African who presented at a tertiary hospital with a one week history of severe generalized fatigue. This was associated with difficulty walking followed by a three days history of difficulty breathing, dysphagia, a sensation of upper airway obstruction and upper gastrointestinal obstruction all in an afebrile state. His past history was remarkable for ophthalmologist follow up for about 5 years (since 2011) for bilateral diplopia and a diagnosis of generalized myasthenia gravis about since one year ago (2015). He was initiated on pyrodostigmine and prednisolone which he stopped 3 months prior to presentation after a sustained remission. He denied any history of hypertension, diabetes, thyroid disorder or any autoimmune disorder. On physical examination he was dyspnoeic with respiratory rate of 34breaths/minute, normal blood pressure of 122/88mmHg, heart rate of 68beats/minute, and temperature of 36.6oc and oxygen saturation in ambient air, SpO2 of 90%.
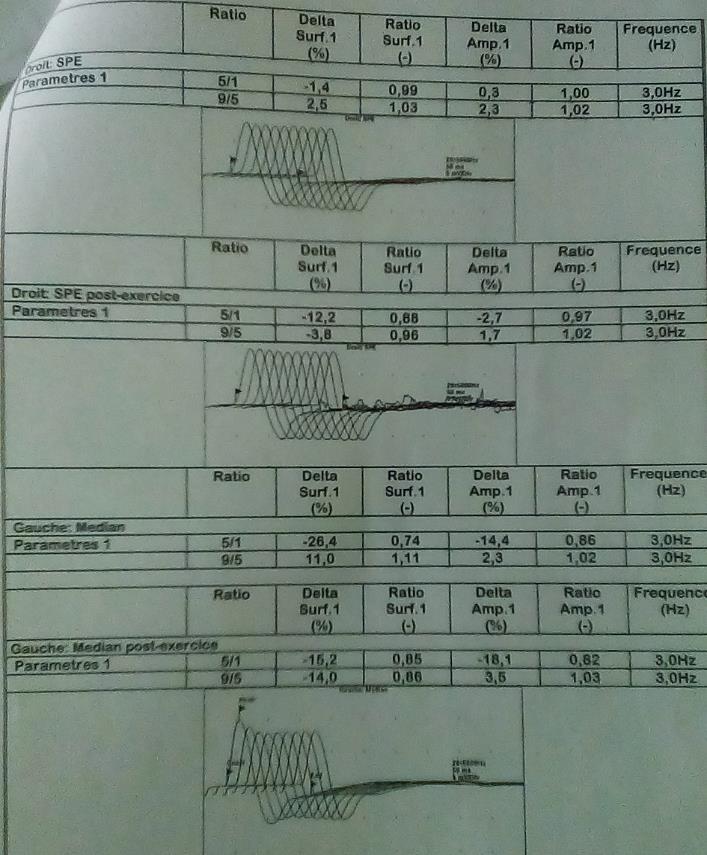
Examination of respiratory system revealed vesicular breath sounds with the following arterial blood gas parameters pH = 7.389, PCO2 = 46.9mmHg, PO2 = 61.2mmHg, HCO3- = 27.7mEq/l. Neurological examination was remarkable for bilateral ptosis, diplopia, generalized muscle weakness, muscle strength decreased (+4/5) in forearm flexors and leg extensors; more marked on the right and easy fatigability of muscles on repeated movement worse on the face, ocular muscles, distal muscles of the forearm and leg. Sensation and motor reflexes preserved. The rest of the clinical assessment was unremarkable. We concluded on a diagnosis of myasthenic crisis (MC) in a patient with generalized myasthenia gravis with precipitating factor non- compliant to medications. Infections as second precipitating factor were to be ruled out. Electromyography done by repetitive nerve stimulation at 3 hertz shows a decrement ranging from 10% - 34% (Figure 1) confirming MG. ABG showed hypoxaemia and hypercapnia (pH = 7.389, PCO2 = 46.9mmHg, PO2 = 61.2mmHg) consistent with respiratory failure. Full blood count, C-reactive protein (CRP), procalcitonine, urea and creatine, serum electrolytes and urine dipstick were normal. Chest-X-ray and chest CT scan done didn’t show evidence of infection, thymomaor thymus hyperplasia. This confirmed our diagnosis of MC precipitated by non-compliant to medications. The patient was hospitalised in the intensive care unit (ICU), intubated and mechanically ventilated. He was placed on intravenous methylprednisolone 120mg 12 hourly, neostigmine 0.25mg/hour (plasmapheresis and intravenous immunoglobulins were not available in our setting) and enoxaparin sodium 4.000IU. The progress was marked by weaning from mechanical ventilation and extubation on day 02 of hospitalization. Respiratory, motor and speech physiotherapies were started after extubation. On day 05 of hospitalisation, he was stable haemodymamically with significant improvement (reduced ptosis no diplopia, no dysphagia. He was transferred from ICU to the neurology unit with continuation of prednisolone, neostigmine and physiotherapy (respiratory, motor and speech). His progress in the neurology unit was uneventful and he was finally discharged from the hospital on day 15 of hospitalisation on the following medications: Pyridostigmine 60mg tablet 6 hourly and prednisolone 20mg tablet 12 hourly.
Discussion
Acquired myasthenia gravis (MG) is a relatively uncommon disorder, with prevalence rates that have increased to about 20 per 100,000 in the US population. MG was first reported in a Native American chief who died in 1664 [9]. The hallmark of the disease is fluctuating weakness and increased by exertion. Weakness increases during the day and improves with rest. Presentation and progression vary. Majority of patients are female. The patients may present with diplopia, dysphagia, ptosis, and limb weakness. Respiratory failure can be a complication during the late course of MG in about 3% to 8% of cases, known as a myasthenic crisis [10]. Our case demonstrates a rare but well known manifestation of MG. Our patient presented 4 years prior to diagnosis (2011) to an ophthalmologist with diplopia compatible with ocular MG that was not diagnosed until 2015 when he presented with generalized fatigue and ptosis to a neurologist and the diagnosis was made. In about 50% of patients, initial presentations are ocular manifestations before becoming generalized [5]. Physical findings can vary in MG as the muscle weakness tends to be more when the muscles are stressed. Muscle strength improves with rest, so the initial physical examination may not reveal any neurological deficit. In our case, there was muscle weakness marked on the right and worsened on repeated movements. The important factor that distinguishes the disorder from other neuropathies is that sensation and reflexes are preserved as in our indexed case. Vital capacity, timed forward abduction, and muscle dynamometry can be used as objective tools to diagnose and assess the disease activity [2]. Respiratory muscle involvement in myasthenia gravis can be of two types: myasthenic and cholinergic crises. The term MC is used to describe muscle weakness because of decreased neuromuscular transmission at the synapse. In contrast, cholinergic crises occur because of excessive depolarisation at the neuromuscular junction. MC can occur as a result of infection, decreased anticholinergic medication, use of aminoglycoside antibiotics, and postoperative stress [2]. Our patient presented with typical features of MC with the precipitating factor being non-compliance to medications as the patient has stopped his medications for three months after a sustained remission and work up for infections were negative.
Diagnostic tests are needed to confirm clinical suspicion include anti-AChR antibodies, electromyography (EMG) repetitive nerve stimulation (RNS) test, edrophonium tests [2]. Associated conditions including thymoma andthymic hyperplasia should be ruled out with appropriate investigations such as chest CT scan [6, 10]. In our patient RNS at 3Hertz done showed a decrement between 10-32% in the muscles groups stimulated. Anti-AChR were not done and chest CT scan and chest X ray done neither showed evidence of thymoma/ thymic hyperplasia no chest infections. Current treatment options for myasthenia gravis include anticholinesterase agents, surgical thymectomy, immunosuppression, and short term but fast acting therapies such as plasma exchange and intravenous immunoglobulin. In general acetylcholinesterase inhibitors agents are used as the first line agents along with surgical thymectomy or immunosuppression [2]. In case of MC admission into the intensive care unit, plasmapheresis or intravenous globulin and steroids are used for induction of remission [11, 12, 13, 14]. In our case, the patient was admitted into the ICU, respiratory support done by intubation and medical management consisted of steroids and acetylcholinesterase inhibitors as described above. The lack of facilities in our setting for plasma exchange and intravenous immunoglobulin hindered their use. This case report highlights the importance of considering neuromuscular disorders in cases of unexplained ocular symptoms / diplopia. Amongst others it also shows how a thorough history taking can help in identification of a precipitant in a patient. Finally, it highlights the place of ICU admission and intubation in the management of MC even in a resource limited setting.
Conclusion
Myasthenia gravis can manifest as ocular symptoms as well as respiratory failure. Albeit the relative rarity of MG in sub Saharan Africa settings, this case highlights the importance of thorough early clinical assessment and investigation of patients with acute ocular symptoms / diplopia.
Declarations
Ethics and consent
Written/ signed informed consent was obtained from the patient for the publication of this cases and accompanying images. Patient’s confidentiality was also maintained throughout. Availability of data and material: Not applicable Competing interests: The authors declare that there is no competing interest Funding: None
Acknowledgement
We express our sincere gratitude to all doctors, nurses and medical students who took part in the management of the patient.
Authors’ contributions
Conception and design of the original manuscript (PNT, CK, RT, GK), Drafting of the manuscript (PNT,RFT, GK), Critical revision of the manuscript for important intellectual content (CK, HNL), Correction of final manuscript (all authors), supervision (PNT, HNL), all authors approved the final version.
References
-
Wendell LC, Levine JM (2011) Myasthenic Crisis. The Neurohospitalist 1(1): 16-22.
-
Vaidya H (2006) Case of the month: Unusual presentation of myasthenia gravis with acute respiratory failure in the emergency room. Emerg Med J 23(5): 410-413.
-
Chaudhuri A, Behan PO (2008) Myasthenic crisis. QJM 102(2): 97–107.
-
Thanvi B, Lo T (2004) Update on myasthenia gravis. Postgrad Med J 80(950): 690-700.
-
Nair AG, Patil-Chhablani P, Venkatramani DV, Gandhi RA (2014) Ocular myasthenia gravis: A review. Indian J Ophthalmol 62(10): 985-991.
-
Elrod RD, Weinberg DA (2004) Ocular myasthenia gravis. Ophthalmol Clin North Am 17(3): 275-309.
-
Rabinstein AA, Mueller-Kronast N (2005) Risk of extubation failure in patients with myasthenic crisis. Neurocrit Care 3(3): 213-215.
-
Sharma S, Lal V, Prabhakar S, Agarwal R (2013) Clinical profile and outcome of myasthenic crisis in a tertiary care hospital: A prospective study. Ann Indian Acad Neurol 16(2): 203-207.
-
Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J (2012) Myasthenia Gravis: A Review. Autoimmune Dis.
-
Sharma SR, Sharma N, Yeolekar ME (2012) An undiagnosed myasthenia gravis presenting as isolated recurrent acute respiratory failure. J Neurosci Rural Pract 3(1): 80-82.
-
Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, et al. (2016) Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med 375(6): 511-522.
-
Pittayanon R, Treeprasertsuk S, Phanthumchinda K (2009) Plasmapheresis or intravenous immunoglobulin for myasthenia gravis crisis in King Chulalongkorn Memorial Hospital. J Med Assoc Thai 92(4): 478-482.
-
Sathasivam S (2011) Current and emerging treatments for the management of myasthenia gravis. Ther Clin Risk Manag 7: 313-323.
-
Kumar R, Birinder SP, Gupta S, Singh G, Kaur A (2015) Therapeutic plasma exchange in the treatment of myasthenia gravis. Indian J Crit Care Med 19(1): 9-13.
- Psychogenic Erectile Dysfunction in Late Adulthood: A Case Report on Clinical Intervention and Intimacy Restoration
- Clinical Trials on COVID-19 in 2025: A New Chapter in Global Health Research
- Innovations and Challenges in Contemporary Medical Clinical Trials: An Editorial Perspective
- Innovations and Challenges in Contemporary Medical Clinical Trials: A Critical Perspective
- Reimagining Clinical Trials: The Power of Continuous Feedback from Medical Reports
- Factors Influencing Brain Drain: Perspectives from a Medical School in Turkey