Neuromielitis Optica Spectrum Disorders (NMOSD) - A Clinical Case
Neuromyelitis optica (sNMO) or Devic’s disease, is a chronic, demyelinating, autoimmune and inflammatory disease of the central nervous system (CNS), that affects predominantly the optic nerve and the spinal cord. Recently unified under a broader term, NMOSD (NMO spectrum disorders), 80% of patients with this disease present detectable serum antibodies, very specific and pathogenomic, that target the water channels aquaporin-4, called AQP4-IgG. Classified as a rare genetic disease with multifactorial origin, NMO affects predominantly females and young adults. The case we present in this study represents an exception. Our patient was diagnosed with NMO at 72, having been previously hospitalized three times, two with transverse cervical myelitis and one with binocular diplopia. These episodes did not leave sequealae and where considered of ischemic origin considering a past cerebro vascular accident. However, the patient presented four more episodes of NMO after his definitive diagnose. In the first two of these attacks, AQP4-IgG testing was requested and positive, with values of 5.31 and 1.62 respectively. No testing for antibodies were requested during his other two attacks. In total, this patient suffered eight attacks in 10 years, which included five attacks with transverse spinal cord myelitis, two with binocular diplopia, and one with dorsal transverse myelitis. The purpose of our paper is to describe this case in detail and to discuss the clinical and neurophysiological testing, genetics, immunology and treatment options available for NMOSD, with the intention of give it more visibility and improve early diagnosis, and considering the long-term debilitating effects that it can have in the patients.
Introduction
Neuromyelitis optica (NMO) or Devic’s Syndrome, was first described by Clifford Albutt in 1870 and characterized by Fernand Gault and Eugene Devic in 1984. It is a chronic, demyelinating, autoimmune and inflammatory disease of the central nervous system (CNS) that affects predominantly the optical nerve and the spinal cord. Described with a multifactorial etiology (genetic, autoimmune, environment) and an estimated range of prevalence of 0.3-4.4/100,000, it is classified as a rare disease [1]. It has been found in all continents and races, but more commonly in non-caucasians (United States, Canada and Europe). NMO can appear at any age range, but predominantly in individuals 30-40 years and in more women than men (ratio 9:1) [2]. Despite being considered a disease of young adults , exceptional cases of onsets during childhood and the elderly have been found [1, 3], including our case study.
Clinically, NMO is characterized by a severe affectation of optic neuritis (NO) and acute transverse myelitis (ATM) or longitudinally extensive transverse myelitis (LETM). The symptoms can appear simultaneously (monophasic) or in phases, with relapses (polyphasic), producing long-term incapacities in young individuals and adults [4]. In 2004, Lennon, et al. [5] reported that most patients with NMO have detectable amount of serum antibodies that target the water channels aquaporin-4 (AQP4), disseminated in the CNS’ astrocytes. These antibodies, termed AQP4-Ab or NMO-IgG are highly specific for clinical diagnosis of NMO, helping to distinguish it from other autoimmune diseases. Therefore, since 2006, the diagnostic criteria for NMO includes AQP4-IgG serology in addition to other laboratory testing, neuroimaging and neurophysiologic testing.
Our Clinical Case
We present here the case of a male with a previous history of arterial hypertension. He was taken into the neurology clinic at 72 years old, with the following clinical presentations: blurry vision, decrease of bilateral visual capacity (agudeza both eyes 2/10), weakness sensation in all four limbs and walking instability. All of the complementary tests performed: blood analysis - including biochemistry, hematogram, coagulation, autoimmune study- and analysis of cerebrospinal fluid - with culture, serology, cytology and viral studies with PCR - resulted in lower than normal ranges and values. The only difference observed were the oligoclonal IgG bands present in the cerebrospinal fluid and the moderate alteration of the hematoencephalic barrier. In the imaging studies, the cerebral MRI showed a thickness and an enhancement of the optic chiasma and the optical tract, clinical manifestations compatible with bilateral optical neuritis (Figures 3a & 3b), whereas the spinal cord MRI revealed small lesions with thickness at C2-C3, typically associated with cervical transverse myelitis. Testing for AQP4-Ab antibodies was negative. However, the patient was diagnosed at his time with NMO taking into consideration his past medical history, with two previous hospitalizations due to relapses of cervical transverse myelitis.
This patient had been hospitalized in three previous times before the definitive diagnose of NMO, twice with idiopathic cervical transverse myelitis and once with binocular diplopia, all without residual symptoms. These episodes were considered to be of ischemic origin due to the patient’s previous history of a cerebro vascular accident (CVA) with severe carotid stenosis. The CVA did not produce any posterior functional limitations. After diagnosis of NMO, the patient presented an additional four relapses. In the first two, the tests for AQP4-IgG were positive, with values of 5.31 and 1.62 respectively; In the other two relapses, this testing was not requested. The imaging tests requested in every relapse were spinal cord MRI, that detected lesions characteristic of myelitis (hyperintensities in T2 and hypointensities in T1, affecting three or more vertebral segments (Figures 1 and 2).
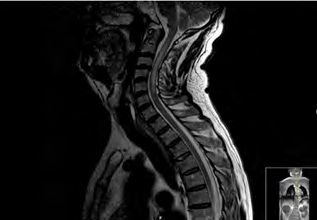
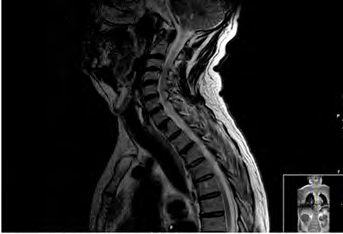
Clinically, the patient has presented variable symptomatology, with motor, sensitive, and sphincter manifestations in the myelitis attacks and visual manifestations during the neuritis optical attacks. Clinically, the myelitis attacks were characterized by nausea, vomiting, sensation of weakness in all extremities, instability walking, decreased strength mainly in the lower left extremity, decreased sensitivity and sphincter alterations (difficulty to initiate micturition, episodes of acute urine retention and constipation). The optic neuritis attacks, also variable, were characterized by diplopia, burred vision and a decreased in bilateral visual acuity.
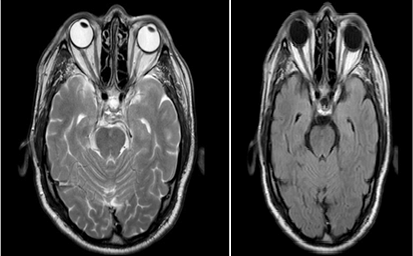
Figure 3a & 3b: Cerebral MRI: Thickening and increased signal in the optic chiasm and optic cintillas.
In total, 5 of the 8 relapses were diagnosed with transverse spinal cord myelitis, 2 with binocular diplopia and bilateral optic neuritis and 1 with dorsal transverse myelitis. All the attacks were treated in an acute way, with pulses of 1 g of methylprednisolone (MP) per day during five consecutive days. The patient responded well and improved clinically during the first three relapses, but the response to steroids was not effective after the fourth attack. At this time, the patient was treated with plasmapheresis therapy in one attack and intravenous immunoglobulins in the others.
After the 8th relapse, it was decided to initiate a preventive treatment with rituximab (anti-CD20 monoclonal antibody) due to the frequency of his relapses (one every fifteen months) and regardless of the immunosuppressive treatment with prednisone and azathioprine already initiated once his definitive diagnosis of NMO was confirmed.
Our patient is currently 81 years old and has not presented any new relapses of neuromyelitis optica. At a functional level he is totally independent, presenting, as his only sequela, a slight weakness in his left lower extremity and paroxysmal painful tonic spasms of short duration (less than one minute), but without any other motor, sensitivity, visual or sphincter dysfunctions. Table 1 summarizes the diagnosis and clinical manifestations for each one of the eight relapses presented by our patient.
| Diagnostic Suspicion | Clinical Presentations | |
|---|---|---|
| Relapse 1(70 years old) | Idiopathic Transverse Spinal Cord MielitisAQP4-IgG: not requested | - Nausea and vomiting |
| Relapse 1(70 years old) | Idiopathic Transverse Spinal Cord MielitisAQP4-IgG: not requested | - Left hemiparesis and right hemihypoesthesia |
| Relapse 1(70 years old) | Idiopathic Transverse Spinal Cord MielitisAQP4-IgG: not requested | - Sphincter dysfunctions (difficulty to initiate micturition, acute urine retention, constipation) |
| Relapse 2(71 years old) | Binocular Diplopia (mistakenly linked to ischemic origin)AQP4-IgG: not requested | - Horizontal diplopia in all directions of the visual field, without evidence of ophthalmoparesis |
| Relapse 3(71 years old) | Idiopathic Transverse Spinal Cord MielitisAQP4-IgG: not requested | - Left hemiparesia, mainly in lower extremity |
| Relapse 4(72 years old) | NEUROMYELITIS OPTICA (NMO)(previous history of spina cord myelitis with current diagnoses of optic neuritis)AQP4-IgG: negative (value < 1) | - Blurred vision and decreased bilateral visual acuity(right eye: 2/10; left eye: 2/10) |
| Relapse 4(72 years old) | NEUROMYELITIS OPTICA (NMO)(previous history of spina cord myelitis with current diagnoses of optic neuritis)AQP4-IgG: negative (value < 1) | - Feeling of weakness in four extremities and unstable walking |
| Relapse 5(74 years old) | Attack of transverse spinal cord myelitis related to NMOAQP4-IgG: positive (value: 5.31) | - Left Hemihypoesthesia and weakness, mainly in upper extremities |
| Relapse 5(74 years old) | Attack of transverse spinal cord myelitis related to NMOAQP4-IgG: positive (value: 5.31) | - Left omalgia |
| Relapse 6(76 years old) | Attack of transverse spinal cord myelitis related to NMOAQP4-IgG: positive (value: 1.62) | - Left hemiparesia and hemihypoesthesia |
| Relapse 7(78 years old) | Attack of transverse dorsal myelitis related to NMOAQP4-IgG: not requested | - Continuous pain at the right hypochondrium |
| Relapse 7(78 years old) | Attack of transverse dorsal myelitis related to NMOAQP4-IgG: not requested | - Left lower hemiparesia |
| Relapse 7(78 years old) | Attack of transverse dorsal myelitis related to NMOAQP4-IgG: not requested | - Sphincter dysfunctions (nocturia, poor urine stream; constipation) |
| Relapse 8(80 years old) | Attack of transverse spinal cord myelitis related to NMOAQP4-IgG: not requested | - Right cervicalgia and omalgia |
| Relapse 8(80 years old) | Attack of transverse spinal cord myelitis related to NMOAQP4-IgG: not requested | - Hemiparesia in right extremities |
Table 1: Description of the diagnostic suspicion and clinical presentations for each one of the eight attacks of neuromyelitis op
Discussion
Neuromyelitis optica (NMO, Devic’s syndrome) is a rare neurological and chronic disease of the central nervous system, characterized by demyelination and inflammation, with attacks of neuritis optica (NO) and acute transverse myelitis (ATM) or longitudinally extensive transverse myelitis (LETM) lesions [4].
In the past, NMO was considered a clinical opticoespinal (Asiatic) variant of multiple sclerosis (MS), wrongly diagnosed in more than 20% of the patients, especially before the discovery of aquaporin-4 antibodies, AQP4-Ab [2, 6]. Our current knowledge of the clinical presentations, laboratory testing, neuroimaging and anatomical pathology allows now for a clear distinction between NMO and MS. An important advance occurred In 2004 with the discovery of detectable serum antibodies, highly specific, that target the water channel aquaporin-4 (AQP4-immunoglobulin G, IgG). Most patients with NMO present these antibodies, which are absent in MS [1]. In 2006, this serological analysis was incorporated into the NMO diagnostic criteria [2, 7]and validated in different ethnic and racial cohorts worldwide. The term “ Neuromyelitis Optica Spectrum Disorders” (NMOSD) was introduced in 2007 as a new nomenclature to facilitate consensus and unify diagnosis criteria, considering the added diagnostic criteria and the diverse clinical manifestations. Additionally, previous diagnostic criteria of NMO only considered lesions in the optic nerve and spinal cord, but today we know that the lesions can extend to other parts of the SNC.
Therefore, under this new classification of NMOD four types of patients are recognized [7]: a. Patients that are AQP4-IgG-seropositive, with limited forms of NMO (e.g., first-attack LETM, NO? recurrent or bilateral optic neuritis) who were at high risk for future attacks. b. Patients with cerebral, diencephalic and brainstem lesions. c. Patients that are AQP4-IgG-seropositive with coexisting autoimmune disorders. d. Patients diagnosed with opticospinal MS (Asiatic).
In 2015, the International Panel for NMO Diagnosis (IPND) was convened to revise the diagnostic criteria for clinical decision-making. A consensus was reached and a report produced with recommendations to unify all the terms under NMO and NMOSD, establishing core clinical characteristics and a simpler further classification of patients based on serological testing: a) Patients with AQP4-IgG and b) Patients without AQP4-IgG or unknown.
The first group, includes patients with 1) at least one core clinical characteristic. 2) positive test for AQP4-IgG. 3) exclusion of alternative diagnoses.
The second group includes a more rigorous diagnostic criteria: patients with 1) Presence of at least two core clinical characteristics, occurring as a result of one or more clinical attacks. 2) Negative test for AQP4-IgG and 3) exclusion of alternative diagnoses.
In this second group, the following requirements should also be met: a) At least one core characteristic must be optic neuritis, acute myelitis with LETM or area postrema syndrome. b) Dissemination in space (2 or more different core clinical characteristics). c) Fulfillment of additional MRI requirements, as applicable (Table 2) [8].
Diagnostic criteria for NMOSD with AQP4-IgG
1- At least 1 core clinical characteristic
2- Positive test for AQP4-IgG using best available detection method (cell-based assay strongly recommended)
3- Exclusion of alternative diagnoses
Diagnostic criteria for NMOSD without AQP4-IgG or NMOSD with unknown AQP4-IgG status
1- At least 2 core clinical characteristics occurring as a result of one or more clinical attacks and meeting all of the following requirements:
a. At least 1 core clinical characteristic must be optic neuritis, acute myelitis with LETM or area postrema syndrome b. Dissemination in space (2 or more different core clinical characteristics) c. Confirmation of additional MRI requirements, as required
2- Negative test for AQP4-IgG using best available detection method, or testing unavailable
3- Exclusion of alternative diagnoses
Core clinical characteristics
1- Optic neuritis
2- Acute myelitis
3- Area postrema syndrome: episode of otherwise unexplained hiccups or nausea and vomiting
4- Acute brainstem syndrome
5- Symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions
6- Symptomatic cerebral syndrome with NMOSD-typical brain lesions Additional MRI requirements for NMOSD without AQP4-IgG and NMOSD with unknown AQP4-IgG status
1- Acute optic neuritis: requires brain MRI showing (a) normal findings or only nonspecific white matter lesions, OR (b) optic nerve MRI with T2-hyperintense lesion or T1-gadolinium enhanced lesion extending over >½ optic nerve length or involves optic chiasm (Figure 1)
2- Acute myelitis: requires associated intramedullary MRI lesion extending over ³3 contiguous segments (LETM) OR ³3 con- tiguous segments of focal spinal cord atrophy in patients with history compatible with acute myelitis.
3- Area postrema syndrome: requires associated dorsal medulla/area postrema lesions
4- Acute brainstem syndrome: requires associated periependymal brainstem lesions
- Abbreviations = AQP4: Aquaporin-4; IgG: Inmunoglobulin G; LETM: longitudinally extensive transverse myelitis lesions;
- NMOSD: neuromyelitis optica spectrum disorders; MRI: magnetic resonance imaging.
Table 2: Diagnostic criteria of NMOSD for adult patients [7].
Etiology
Similar to other autoimmune diseases, NMO has a multifactorial origin, with influence of infectious, environmental and genetic factors. Several studies have found that in up to 20-30% of cases, NMO attacks are preceded by an infection, such as flu, vaccination or pregnancy (during the last trimester) and postpartum period). No cases of direct transmission mother-to-fetus has been found [1]. In 2017, Vieira et al reported the case of a 9-year-old girl admitted initially with fever and headache, and later with ataxia and bilateral loss of vision. Cerebral and spinal cord MRI revealed changes in the posterior optic nerves and optic chiasm, optic tracts and lateral genicular bodies, with three small lesions in the posterior columns of the dorsal spinal cord at T2, T4- T5 and T6 levels. Serological testing was positive for Borrelia burgdorferi IgM. The test for anti-AQP4 was negative and for anti-myelin oligodendrocyte glycoprotein antibody (MOG) was positive. After the adequate treatments, the young patient slowly recovered, MRI was normal and MOG assay became negative. It was concluded that she had suffered spinal cord and optic nerve demyelination, anti-MOG mediated, as a response to the infection by the bacteria [9].
Genetics
NMOSD has been reported in different populations around the world, but it seems more frequent in non- Caucasians. The clinical phenotype differences between populations indicate a multifactorial origin, with strong effect of genetic susceptibility over other factors (environmental, infectious). Epidemiological findings suggest the involvement of several genes (polygenic), such as those involved in the synthesis of the T cell beta chain receptor (TCRB gene), the immunoglobulin variable chain protein (VH2-5 gene), the myelin basic protein (MBP gene), the cytotoxic T-lymphocyte associated protein (CTLA-4 gene) and the interleukin-1 protein (IL1B gene).
In 2018, Alonso, et al. [10] performed a genetic study of 35 patients with NMO in a Mexican population and they found a higher presence of HLA-DBR1 alleles (for human leukocyte antigen) in individuals with NMO and MS. The HLA complex consist in a family of genes that helps the immune system distinguish the body’s own proteins from foreign invaders (viruses and bacteria). The HLA-DRB1 gene, located in chromosome 6 at position 21.32, belongs to a group of MHC (major histocompatibility complex) genes called MHC class II, which provide instructions for making surface proteins. These proteins attach to peptides outside certain cells. MHC class II proteins display these peptides to the immune system, and if recognized, it triggers an attack to the invader. Specifically, the protein produced by DRB1, a beta chain, binds to another protein, an alpha chain, produced by the HLA-DRA gene, to form a functional protein complex, the HLA-DR.
HLA-DRB1 gene has been associated with many autoimmune disorders, and hundreds of different versions (alleles) have been identified. In their study, Alonso et al. found a higher frequency of HLA-DRB1*03 and HLA-DRB1*10 alleles in patients with NMO than in controls. Additionally they found no differences in the other alleles that have been described in MS subjects (HLA-DRB1*04, HLA-DRB1*08 and HLA-DRB1*13)
Clinical Presentations and Evaluation
Neuromyelitis optica is a disease more prevalent in young adults (30-40 years of age), with median age at onset of 39 years, and affecting more women than men (ratio 9:1) [2]. However, in exceptional cases it can affect children and elderly, as in the case of our patient [1, 3].
Clinically, this disease involves optic neuritis (ON) and acute transverse myelitis (ATM) , in a polyphasic course (with attacks and remissions) in 60% of the cases in one year, and in 90% of the cases in three years, as in the clinical case we present here. Monophasic forms with ON and ATM simultaneously or within 30 days or less, are less frequent. In some sporadic cases, NMOSD was associated with dysphagia [11].
Optic Neuritis is characterized by the loss or decrease of visual acuity, pain during ocular movement and dyschromatopsia (difficulty to distinguish or perceive colors, also known as Daltonism) in one or both eyes. Generally, ON starts unilateral and becomes bilateral in a short period of time. The visual field presents a central scotoma, or blind spot, and the fundus of the eye can be normal or pathological (with edema, atrophy or pallor of the optic papilla or optic disc). Blindness can occur as a sequelae in 60% of the recurrent disease forms and in 22% of the monophasic.
Acute transverse myelitis occurs, in most cases, months or years after optic neuritis, but in monophasic courses and frequently in young adults, it can appear after only a few days. ATM involves the inflammation and affectation of three or more vertebral segments, causing lesions which extend in a longitudinal pattern, known as LETM (longitudinal extensa transverse myelitis). The areas of the spinal cord most commonly affected are the upper cervical and dorsal segments. This produces disturbances at the motor level (with loss or decrease of strengh), at a sensitive level (with paresthesias or dysesthesias) and alterations in sphincter functioning. Paintful Paroxysmal Tonic Spams (PTS) are also recurrent [12]. It has been found that radicular pain, paintful PTS with a duration of 20-45 seconds) and the Lhermitte’s sign (sudden sensation of “electric shock” that passes down to arms and legs during cervical flexion) can appear in 33% of the recurrent forms. Although the lesions found in the CNS do not affect the white matter nor produce encephalic syndromes, the damage of the brainstem and the spinal cord can produce incoercible and intractable hiccups, nausea and vomiting, even respiratory failure as a consequence, and which represents the most frequent cause of death for these patients [1].
The Neuropathologic characteristics of NMOSD include deposits of serum antibodies and complement, loss of astrocytes, secondary degeneration of oligodendrocytes and neurons, and necrotic lesions with infiltration of, neutrophilic and eosinophilic granulocytes [13].
Neuroimaging: MRI and OCT
Magnetic resonance imaging (MRI) and Optic Coherence Tomography (OCT) are indispensable techniques in the evaluation and differential diagnosis of NMOSD [14]. Detection of spinal cord lesions extending for more than 3 segments, often with irregular enhacement contrast during weeks or months, and central necrosis and cavitation less frequently, are very characteristic and distinctive of NMOSD. Despite their appearance, most of these spinal cord lesions are treatable and could heal completely. New discoveries about the appearance of cerebral lesions during NMO, demonstrated that in contrary to what was initially thought, the majority of the patients, up to 60%, although they might be clinically silent, they could present brain lesions mainly unspecific and detectable as hyperintensities in their MRI. In some cases, individuals with NMOSD showing these brain lesions were considered diagnostic of Multiple Sclerosis (MS). Therefore, it is currently accepted that although many patients might present normal results at the beginning of the disease, brain lesions, including those similar to MS-lesions, do not exclude a diagnosis of NMO and should be considered a red flag.
Recent MRI studies [15] revealed differences between NMOSD and MS cortical and white matter lesions: In NMO, cortical lesions are absent and white matter lesions are subcortical and lack central venule (unlike MS, with periventricular plaques with central venule). Additionally, brain lesions in NMO are generally located in areas of high expression of aquaporin-4 expression, such as the diencephalon, hypothalamus and corpus callosum. Contrast enhancement images in the cerebral MRI with cloud-like shapes and ependymal enhancement pencil-like are also characteristic of NMO.
Optical coherence tomography is becoming a very useful tool in neuroimmunological studies, and its utility to distinguish NMO and MS from other inflammatory conditions with ocular involvement has been investigated in recent years. It is a non-invasive technique for visualization of the unmyelinated CNS axons within the retina, the retinal nerve fiber layer (RNFL), and their neurons, the ganglionic cells of the retina. In the case of NMO, the loss (thinning) of the retina is associated to the clinical attacks of optic neuritis, and one single acute attack of optic neuritis can cause more damage to the RNFL in NMO than in MS. In a study by Ringelstein, et al. [16] it was found that 12.3% of the patients presented reduced amplitudes of their visual evoked potentials, 41.9% presented prolonged latencies and 14% presented lack of response. Delayed P100 latencies in eyes without prior neuritis optica suggested a subclinical affection in patients presenting longitudinally extensive transverse myelitis lesions (LETM) with no history of clinical NO.
Autoimmune Specificity
Aquaporins (AQP) are a type of transmembrane proteins distributed in many organs throughtout the human body, with maximum concentrations in the central nervous system (cerebral and cerebellar cortex, posterior segment of the optic nerve, and Muller cells of the retina, ependyma, hippocampus and spinal cord) and the renal medulla. There are 13 known mammalian AQP, with AQP-4 being highly characteristic of the CNS, mainly expressed in the astrocytes (in the processes in contact with the blood vessels) and particularly concentrated in the pial surface of the brain and the ependymal surfaces in contact with the cerebrospinal fluid. AQP-4 is related mainly with the regulation of transmembrane water fluidity. Besides its structural features, the relative permeability in certain regions of the blood-brain barrier (BBB) determines a higher vulnerability to IgG AQP4 antibodies, such as in the case of the prelaminar region of the optic nerves. At the spinal cord level, a higher concentration of AQP is found in the fibrous astrocytes of the two superficial lamina of the posterior processes of the spinal cord. The distribution of the ependyma and its higher permeability supports the appearance of periventricular and periependymal lesions in this region. The discovery of the relevance of antibodies to AQP4 in patients with NMOSD was essential to redefine the clinical spectrum of this disease. AQP4-IgG are detected at a higher percentage at the beginning of the disease, during a first attack (NMO limited). After this first attack of LETM and/or NO with IGg-NMO positive, the risk of recurrence and conversion to definitive NMO is high [1].
The permeability of the BBB increases during systemic inflammatory processes, and some studies have described correlations between infectious processes (viral), immunizations and attacks of NMO. In the study by Jarious, et al. [6] of 175 patients, it was found that 29% of the seropositive patients (with antibodies) had history of infectious processes, compared to 18% of the seronegatives (without antibodies). In this same study it was also found that these patients presented contrasting disease patterns: Those with antibodies were predominantly female, with systemic inflammatory episodes, more severe attacks, higher visual loss during NO, higher frequency of motor sintomas and more extensive spinal cord lesions. Patients without antibodies presented more frequently bilateral NO, with or without myelitis, and monophasic course of disease.
This results supports the hypothesis of the presence of a different autoantigen in these two types of patients, and suggest that seronegatives have a different underlying pathogenesis. Technological advances in AQP4-IgG have improved the diagnosis sensitivity without compromising specificity. The International Panel for the diagnosis of NMO recommended testing with cell-based serum assays (flow cytometry-based detection or microscopy) because they optimize autoantibody detection and have high sensitivity and specificity [7]. In 2015, Omar, et al. [8] described a case study with 73% sensitivity and a 91% sensitivity for NO [8] and other indirect methods, such as ELISA and immunofluorescent assays have lower sensitivities and might yield false positive results.
AQP4-Ab antibodies are considered pathognomonic and are detectable in approximately 80% of the patients with NMO, helping to distinguish this rare disease from multiple sclerosis (MS) [13]. However, 20-30% of the patients are seronegative for AQP4-IgG. Additionally, in
20% of these seronegative individuals, detectable levels of another antibody for a different type of protein, myelin oligodendrocyte glycoprotein (MOG), have been found. Yet, these patients present similar clinical manifestations of NMO, NO and transverse myelitis [17].
Several studies have found that the antibodies detected in a higher frequency are the antinuclear (43,8%) and the anti-SSA/Ro and anti-SSB/La (anti- Sjögren’s syndrome- related antigens A and B respectively) (15,7%). NMO with positive anti-AQP4 can be associated with other autoimmune diseases, some organ specific, such as miastemia gravis and autoimmune tiroiditis and some non-organ specific, such as systemic lupus erythematosus (SLE) [18] and Sjögren Syndrome (SS) [1, 19]. Rare associations between NMO and celiac disease [20] and between NMO and amyotrophic lateral sclerosis (ALS) have also been described [21]. These findings support the hypothesis of the existence of common neuroinflamatory pathological processes.
Therapy
The main two treatment goals for NMO, considering that it is a syndrome with attacks, are first, to improve the relapse- associated symptoms of NO and/or LETM, and secondly, to stabilize the disease course long-term and to prevent new relapses. It is important to clarify that there are not enough prospective clinical studies with high levels of evidence, so these treatments are based on recommendations by experts and mostly based on retrospective case reports.
The most recommended treatment of acute disease attacks consist in the use of a five consecutive days course of 1 g methylprednisolone (MP) per day. 80% of the patients respond well to this treatment. If the condition does not sufficiently improve and the attacks are too acute or lack response to steroids, therapeutic plasma exchange (TPE) can be performed alone or in combination with MP. In this case, improvement was noted in about 50-60% of patients. Intravenous immunoglobulins (IVIg) have also been used for acute relapses and no response to steroids, and despite some discussions about their primary role, it is a safe therapy, well tolerated by most patients and could be an alternative treatment for patients with relapses or with contraindications (hypersensitivity for example) for other therapies [22].
The long-term preventive treatment consists mostly in the use of immunosuppressors and should be initiated as early as possible. It is recommended for patients with many relapses or for those with a high risk to develop a recurrent disease. The combination of azathioprine (AZA) and MP is the most used. AZA is also currently the first-line therapy to prevent attacks of NMO and to avoid the long- term side effects of corticosteroids. Studies show that when started early in patients with NMOSD, AZA is effective, well tolerated and improves neurological functionality in the EDSS scale (Expanded Disability Status Scale) and the visual score. Another immunosuppressor being prescribed is mycophenolate mofetil (MMF). This treatment is often given to patients with contraindications or side effects to AZA, such as induced toxicity due to low thiopurine methyltransferase activity [23].
Biological therapies are starting to be used more frequently now, in addition to the classic immunosuppressors described above. B-cell depletion with rituximab (RX) and other agents, inhibition of interleukin 6 (IL-6) receptor with tocilizumab, and blocking of complement component 5 with monoclonal antibody eculizumab, are some examples of these new promising treatments [24].
Several clinical cases have shown that treatment with RX is an effective first-line therapy in NMO/NMOSD. This monoclonal antibody (anti-CD20) is also being used in patients who have not responded to previous immunosuppressive therapies, such as AZA. However, there is still great heterogeneity in the treatment and multiple factors to consider: the required vaccines before and after treatment; the number, frequency and dosing of the regimens; the prevention of adverse reactions to infusion; and the prevention of infections during treatment [25].
Final Remarks
The differential diagnosis of NMOSD in the clinical practice is often difficult, regardless the phenotypic and serological characteristics of the disease, due to the following factors: 1) Several diseases with autoimmune etiologies, infectious or neoplastic can simulate NMOSD phenotypes. 2) Patients with NMOSD might present limited clinical manifestations, especially in the early stages of the disease. 3) The results of testing for AQP4-IgG can be affected by several factors, such as the assay method, the serological phase, the stage of the disease or the types of treatments being used. 4) Some patients with NMOSD are seronegative for AQP4- IgG. 5) Results from testing for AQP4-IgG might not be available.
It is crucial to understand that despite the similarities, different phenotypes are possible in NMOSD, each with its own and characteristic pathogenesis, prognosis and more importantly, necessary treatment. Awareness of the clinical, serological, radiological and detailed prognosis of these disease by the family doctors will improve the adequate diagnosis and intervention of these patients [26, 27].
Therefore, it is clearly necessary to include NMOSD in the differential diagnosis for patients with findings compatible with demyelinating diseases, considering that an early diagnose and a preventive therapy with immunosuppresor drugs as soon as possible could improve their prognosis and their quality of life, reducing the possibility of becoming disable in their lives due to a residual effect of the attacks.
References
-
Edgar Carnero Contentti, Felisa Leguizamón, Pedro Ernesto Colla Machado, Alonso R (2013) Neuromielitis óptica: actualización clínica y terapéutica. Sociedad Neurológica Argentina. Elsevier, España.
-
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, et al. (2014) Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 261(1): 1-16.
-
Castilla Guerra L, Fernández Moreno MeC, Carmona González M, Alvarez Suero J (2012) Neuromyelitis optica in the elderly: An unusual demyelinating disease. Rev Esp Geriatr Gerontol 47(3): 134-135.
-
Chiquete E, Navarro-Bonnet J, Ayala-Armas R, Gutiérrez- Gutiérrez N, Solórzano-Meléndez A, et al. (2010) Neuromielitis óptica: actualización clínica. Rev Neurol 51: 289-294.
-
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, et al. (2004) A serum antibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364(9451): 2106-2112.
-
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, et al. (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation 9: 14.
-
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, et al. (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85(2): 177-189.
-
Tomás Omar ZB, Antonio VC, Juan Pablo VQ, Cristian Fernando VZ, María Fernanda VH, et al. (2015) Neuromielitis Óptica De Devic: Reporte De Caso En Popayán. Medicina (Bogotá)Diciembre, pp: 376-384.
-
Vieira JP, Sequeira J, Brito MJ (2017) Postinfectious Anti- Myelin Oligodendrocyte Glycoprotein Antibody Positive Optic Neuritis and Myelitis. J Child Neurol 32(12): 996- 999.
-
Alonso VR, de Jesus Flores Rivera J, Garci YR, Granados J, Sanchez T, et al. (2018) Neuromyelitis Optica (NMO IgG+) and Genetic Susceptibility, Potential Ethnic Influences. Cent Nerv Syst Agents Med Chem 18(1): 4-7.
-
Cousins O, Girelli E, Harikrishnan S (2019) Neuromyelitis optica: an elusive cause of dysphagia. BMJ Case Rep 12(1).
-
Carnero Contentti E, Leguizamón F, Hryb JP, Celso J, Pace JL, et al. (2016) Neuromyelitis optica: association with paroxysmal painful tonic spasms. Neurologia 31(8): 511-515.
-
Pache F, Wildemann B, Paul F, Jarius S (2017) Neuromyelitis optica. Fortschr Neurol Psychiatr 85(2): 100-114.
-
Akaishi T, Nakashima I, Sato DK, Takahashi T, Fujihara K (2017) Neuromyelitis Optica Spectrum Disorders. Neuroimaging Clin N Am 27(2): 251-265.
-
Jarius S, Wildemann B, Paul F (2014) Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol 176(2): 149-164.
-
Ringelstein M, Kleiter I, Ayzenberg I, Borisow N, Paul F, et al. (2014) Visual evoked potentials in neuromyelitis optica and its spectrum disorders. Mult Scler 20(5): 617- 620.
-
de Seze J (2019) Myelin oligodendrocyte glycoprotein antibodies in neuromyelitis optica spectrum disorder. Curr Opin Neurol 32(1): 111-114.
-
Chiganer EH, Hryb JP, Carnero Contentti E (2017) Myelitis and Lupus: Clinical Manifestations, Diagnosis and Treatment. Review. Reumatol Clin 13(6): 344-348.
-
Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, et al. (2008) Neuromyelitis optica and non organ- specific autoimmunity. Arch Neurol 65(1): 78-83.
-
Díaz Díaz A, Hervás García M, Muñoz García A, Romero Santana F, Pinar Sedeño G, et al. (2017) Celiac disease and neuromyelitis optica: A rare but possible association. Neurologia 34(8): 547-549.
-
Li A, McGranahan T, Su E, Kipp L, Gold CA (2019) Coexistence of Neuromyelitis Optica and Amyotrophic Lateral Sclerosis: A Case Report. Neurohospitalist 9(1): 37-40.
-
Magraner MJ, Coret F, Casanova B (2013) The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 28(2): 65-72.
-
Edgar Carnero Contentti, Javier Pablo Hryb, Jose Luis Di Pace, Edson Chiganer, Mónica Perassolo (2013) Rol de la detección de los anticuerpos anti-acuaporina 4 (IgG- NMO) en el espectro de la neuromielitis óptica. Sociedad Neurológica Argentina: Elsevier España 5(2): 108-113.
-
Kleiter I, Gold R (2016) Present and Future Therapies in Neuromyelitis Optica Spectrum Disorders. Neurotherapeutics 13(1): 70-83.
-
Ciron J, Audoin B, Bourre B, Brassat D, Durand-Dubief F, et al. (2018) Recommendations for the use of Rituximab in neuromyelitis optica spectrum disorders. Rev Neurol (Paris) 174(4): 255-264.
-
Kim SM, Kim SJ, Lee HJ, Kuroda H, Palace J, et al. (2017) Differential diagnosis of neuromyelitis optica spectrum disorders. Ther Adv Neurol Disord 10(7): 265-289.
-
Alejandro N Antezana Siles, Rodrigo A Vallejos, Antezana AO (2016) Longitudinal transverse myelitis: similar presentations, different etiologies. Gac Med Bol, pp: 99- 102.
- Psychogenic Erectile Dysfunction in Late Adulthood: A Case Report on Clinical Intervention and Intimacy Restoration
- Clinical Trials on COVID-19 in 2025: A New Chapter in Global Health Research
- Innovations and Challenges in Contemporary Medical Clinical Trials: An Editorial Perspective
- Innovations and Challenges in Contemporary Medical Clinical Trials: A Critical Perspective
- Reimagining Clinical Trials: The Power of Continuous Feedback from Medical Reports
- Factors Influencing Brain Drain: Perspectives from a Medical School in Turkey