Clinical, Biochemical and Radiological Features of an Infant Affected by Leigh Disease - A Case Report
Leigh disease, also known as subacute necrotising encephalomyelopathy (SNE), is a rare inherited progressive neurometabolic life threatening mitochondrial disorder. Early diagnosis and early treatment will help in reducing both morbidity and mortality associated with this disorder. We report a 2.5 months old Indian infant with clinical, biochemical and radiological features of Leigh Disease. This is the first case report of a child with Leigh Disease of early onset
Introduction
Leigh disease is a rare inherited progressive neurometabolic, life threatening mitochondrial disorder. It has extensive clinical, biochemical, neuroimaging and genetic heterogeneity. Prevalence at birth is 1:40000[1]. It is transmitted as autosomal recessive, X- linked recessive or as maternal inheritance. It is caused by mutations in nuclear genes or mitochondrial genes[2].
Case Presentation
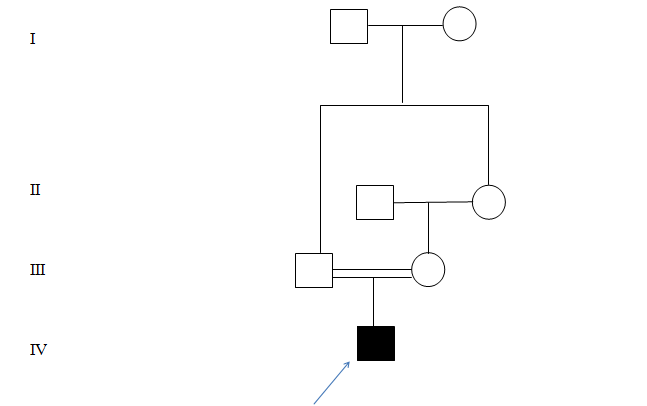
A two and a half month old male child, 1st born to 2nd degree consanguineously married couple as shown in figure 1 with uneventful perinatal history, presented with history of fever which subsided after 4 days, followed by excessive cry, apnoeic spells and excessive arching of the body since 2 days. There was no history of convulsions. Development was appropriate for age. Family History: There was no history of abortion or still birth. No history of similar illness in the family.
Examination The baby's weight was 4.5kg (3rd -50th percentile), length was 58cm (3rd -50th percentile) and occipito frontal circumference was 38cm (3rd -50th percentile). He was pink, euthermic with no dysmorphic features. He had excessive cry and was not consolable. He had long inspiratory pause almost lasting for 30-40 seconds. His pulse rate was 127/ min (120-160), respiratory rate was 44/min (20-40) and oxygen saturation was 77% (>92) @ Room Air. His capillary refill time was < 3 seconds (<3).
Central Nervous System Examination
Baby was lethargic. Cranial nerves examination was normal. There was hypertonia in both the lower limbs. Power was 3/5 in all limbs. Deep tendon reflexes were exaggerated with bilateral positive babinski sign. Pupils were dilated and sluggishly reacting to light. Choreoathetosis was noted. Newborn reflexes like sucking and rooting reflexes were present. Fundus examination was normal. Other systems: Gaseous distension was present per abdomen. His cardiovascular and respiratory systems were normal. Provisional Diagnosis 1. Neurometabolic disorder Table 1: Arterial blood gas analysis
2. Krabbe disease 3. Sepsis 4. Meningitis 5. Kernicterus The baby was investigated further
Investigations
Routine hemogram revealed Hemoglobin 9g% (9-14). Total Leucocyte Count was 9900 cells per mm3 (5000- 10500). Differential count of P30L70 (L>P). Platelet count was 835000 cells per mm3 (150000-400000). Peripheral smear showed normocytic normochromic blood picture with reactive thrombocytosis. Prothrombin time was 16.8seconds (control 15), partial thromboplastin time was 30.9 seconds (control 30). Random plasma glucose was 85.1mg/dl ( 60-100), Urea was 26mg/dl (10-36), Creatinine was 0.7mg/dl (0.2-0.4), ionized calcium was 5.1 mg/dl (4.8-4.92), serum C-reactive protein-12.8mg/L (0.8-15.8), cerebrospinal fluid c-reactive protein was < 6mg/L(0.8-5.8), Cerebrospinal fluid analysis showed 3 cells, all lymphocytes with normal glucose and protein levels. Arterial blood gas (ABG) analysis showed metabolic acidosis with high anion gap as shown in table 1. Plasma ammonia was low. Arterial blood lactate and CSF lactate were high as shown in Table 2.
| Result | Normal Value | Interpretation | |
|---|---|---|---|
| pH | 7.304 | 7.35-7.45 | Acidosis |
| pCO2 | 20.3 mmHg | 35-45 | Low |
| pO2 | 100.7mm Hg | 80-100 | Normal |
| SO2 | 96.30% | 80-100 | Normal |
| HCO3- | 9.9mEq/L | 21-28 | Low |
| BE | -15.1mEq/L | -7to +1 | Low |
| Na+ | 134mEq/L | 135-145 | Low |
| K+ | 4.4mEq/L | 3.5-6 | Normal |
| Cl- | 100mEq/L | 98-106 | Normal |
| Anion Gap | 24.1mEq/L | 7-16 | High |
| Metabolic acidosis with high anion gap. |
Table 1: Arterial blood gas analysis
| Norma | l | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Test | Result | I | nterpretation | ||||||||
| Value | |||||||||||
| Plasma Ammonia | 13 µg/dl | 18-74 | low | ||||||||
| Lactate ( Arterial Blood) | 32.9mg/dl | 3.6-18 | high | ||||||||
| Lactate ( CSF ) | 26.9 mg/dl | 5.4-20 | high |
Table 2: Special biochemistry tests.
Liver function test showed mild derangement with aspartate transaminase 97 IU/L (15-55), alanine transaminase 62 IU/L (5-45). Blood culture and CSF culture showed no growth.ECG and Chest X Ray were normal.
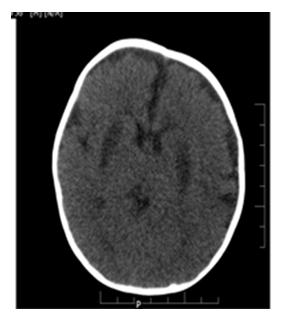
CT Brain (Plain ) as shown in figure 2, at the level of basal ganglia, showed bilateral symmetrical hypodensities noted in the putamen.
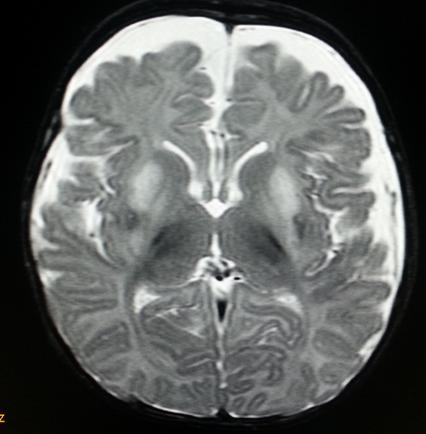
MRI Brain showed a. Symmetric abnormal signal intensities seen involving bilateral globus pallidi and putamen appearing hyperintense on T2W1, hypointense on T1W2 and FLAIR sequences. b. The involved areas showed diffusion restriction with matched Apparent Diffusion Co-efficient hypointensity. c. Diffuse frontotemporal atrophic changes are seen as evidenced by widening of CSF spaces and prominent basifrontal and basitemporal cisterns. d. Degree of myelination appears adequate for patient’s age MRI Brain showed features suggestive of Leigh disease as shown in Figures 3 and 4.
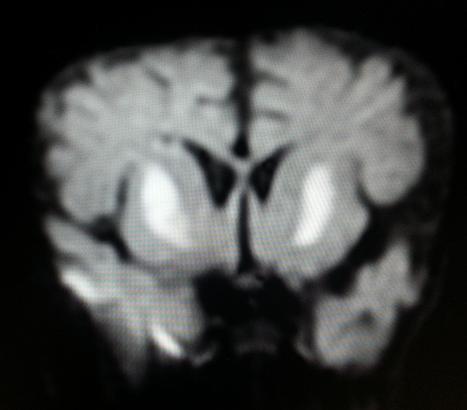
Differential diagnosis: Hypoxic ischemic encephalopathy, metabolic encephalopathy. Final Diagnosis: Leigh disease Treatment: The baby was started on antibiotics, anticonvulsants, thiamine, biotin, carnitine and supportive therapy including mechanical ventilation for impending respiratory failure. The baby improved over the next four days and was later extubated. Post extubation baby remained hemodynamically stable. Outcome: The baby improved.
Discussion
The criteria for diagnosis of Leigh disease are progressive neurological disease with motor and intellectual developmental delay, signs and symptoms of brainstem and / or basal ganglia disease, raised lactate in blood and / or CSF, characteristic bilateral symmetric necrotic lesions in the basal ganglia and / or brain stem. Neuroimaging[3]: MRI plays a very important role in the diagnosis of SNE. The most characteristic neuroradiological findings are bilateral symmetrical focal hyperintensities in the basal ganglia, thalamus, substantianigra and brainstem at various levels on T2W1 MRI. In the basal ganglia, the putamen is particularly involved. Low attenuation in the putamen on CT is considered to be characteristic of the disease. MR Spectroscopy shows elevated lactate and choline and depressed N-acetyl aspartate. Specific therapy for Leigh syndrome is not available. The aim of supportive therapy is to improve ATP production and reduce lactate levels. Thiamine is reported to improve the neurological status in some children. Marked improvement with riboflavin was observed which nearly normalized the ATP production. Use of intravenous soyabean oil resulted in rapid clinical and biochemical improvement in children with acute central respiratory failure. Ketogenic diet is found to be useful in children with pyruvate dehydrogenase complex deficiency. Coenzyme Q and carnitine have also found to be effective. Intramuscular botulinum toxin is found to provide relief from dystonia in children having dystonia[4]. Pronuclear transfer ( PNT) and maternal spindle transfer ( MST ) are two invitro fertilization (IVF) based techniques which seem to have the potential to prevent transmission of maternally inherited Leigh disease caused by mutations in the mitochondrial genes[5]. Egg donation, egg donation with surrogacy or adoption can be an alternative for Leigh disease caused by mutations in the nuclear genes. This baby presented to us at 2.5 months with excessive cry, breathing difficulty, arching of the body and apneic spells which pointed towards a neurometabolic disorder. ABG showed metabolic acidosis with high anion gap. Blood and CSF lactate were elevated. CT Brain (plain) showed features suggestive of Leigh disease which was confirmed by MRI Brain. This baby was treated with antibiotics, thiamine, riboflavin, carnitine, biotin and supportive therapy including mechanical ventilation for impending respiratory failure which was considered to be factors contributing to his improvement. Presently, the baby is 2.5 years old. He is recovering from the acute episode. He is able to walk with support. His developmental quotient is 13 months. Nuclear gene study has been done for this baby. Mutation was not found. Mitochondrial gene study from muscle biopsy specimen is planned. To our knowledge, this is the first case detected at an early age of 2.5months with clinical, biochemical and radiological features of Leigh disease.
Respiratory chain, pyruvate dehydrogenase complex and pyruvate carboxylase enzymes assay, muscle biopsy and mitochondrial gene mutation analysis could not be performed due to paucity of facilities and financial constraints.
Conclusion
Any child who presents to us with history of lethargy, weakness, convulsions, abnormal breathing, abnormal movements and encephalopathy should be investigated further with ABG, electrolytes, arterial blood and CSF lactate. If CT Brain shows evidence of brain stem or basal ganglia hypodensity, it should be confirmed by MRI Brain to establish the diagnosis of Leigh disease. The child must be treated with mega doses of vitamins and care must be taken to avoid stressful situations and infection.
Acknowledgements
We acknowledge Dr. Navin A Patil and Dr. Kunal Naidu, Department of Radiology MG diagnostics, Bengaluru, Karnataka, India for confirming the clinical diagnosis with appropriate neuroimaging. We acknowledge our Director, Dr. Asha Benakappa, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India for constant encouragement and support.
References
-
Kartikasalwah AL, Ngu LH (2010) Leigh syndrome: MRI findings in two children Biomed Imaging Interv J 6(1): e6.
-
Richard E Behrman (2016) Nelson’s Text Book Of Paediatrics 20th Edition. Saunders Ltd, Pennsylvania pp. 2902.
-
Shrikhande, Piyush K, Aarif MMS, Kunal A, Gurmeet S (2010) A rare mitochondrial disorder: Leigh syndrome - a case report. Italian Journal of Pediatrics 36: 62.
-
Chae-Woo Chung, Suhng-Hee H, Young-Chul C, Young- Ho S, Jin-Soo K, et al. (1990) A Case of Leigh’s Disease with Initial Manifestation of Dystonia. Yonsei Medical Journal 31(2): 274-279. **5.** Po-Cheng H, Huei-Shyong W (2007) A previously undescribed leukodystrophy in Leigh syndrome associated with T9176C mutation of the mitochondrial ATPase 6 gene. Developmental Medicine & Child Neurology 49(1): 65-67.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty