Pseudohypoparathyroidism 1a A New Case Report
Patients with Pseudohypoparathyroidism 1a (PHP-1a) have a typical phenotype, described for the first time by Albright, and therefore called Albrights Hereditary Osteodistrophy (AHO). It is characterized by a round face, flat, wide and low nasal bridge, short neck, obesity, short stature, subcutaneous calcifications and skeletal abnormalities (genu valgum, cubitus valgus, brachydactyly of the 4th and 5th fingers). We presented a case of a 2-month old boy with coarse face (macroglossia, flat, wide and low nasal bridge, pre-auricular pitts), disproportionately short lower limbs and umbilical hernia. Blood test showed TSH :11.7U/mL, Parathyroid hormone (PTH) was 329.20pg/mL, serum calcium 10.3mg/dL and phosphorus 7.1mg/dL. The diagnosis was confirmed with the identification of an inactivating mutation of the GNAS gene on the 20 chromosome (c.1096G>A). We started treatment with oral calcium, calcitriol and levothyroxine. Because of recurrent intense headache, with multiple emergency department visits, he underwent computer tomography that revealed typical images of basal ganglia calcification. Currently with 15 years old, he has typical AHO phenotype.
Case Presentation
A 2-month old Caucasian boy is reference to the pediatric endocrinology consultation after a positive neonatal screening for hypothyroidism.
He was the first born of a non-consanguineous healthy parents. Pregnancy was monitored and uneventful and he was born at 40 weeks by cesarean delivery. He had an Apgar score of 6/9 by the 1st/5th minute, improving with positive pressure mask inflation, but still maintaining nasal flaring and difficulty breathing so he was admitted to de neonatology intensive care unit, with gradual improvement. During the first 24 hours of life he also had hypoglycemia requiring intravenous fluid therapy. He was a macrosomic and macrocephalic newborn (4340g weight SDS +1.86; 50 cm length SDS +0.06; 39cm head circumference, SDS +3.57) with a coarse face (macroglossia, flat, wide and low nasal bridge, pre- auricular pitts), disproportionately short lower limbs and umbilical hernia. Investigations for Beckwith-Wiedemann syndrome and achondroplasias, were negative. On the neonatal screening, performed on the 6th day of life, hypothyroidism was first detected (TSH 22.4 µU/mL, reference <20µU/mL), with transient normalization by the 4th week (TSH 9.6µU/mL, reference <20µU/mL). He repeated by 2 months of age (TSH 11.7µU/mL, reference 0.3-3.8µU/mL; T4L 5.8µg/dL, reference 6.5-17.0µg/dL), and started treatment with levothyroxine with good clinical and analytical response. During follow-up visits, it was notorious a psychomotor retardation, an excessive weight gain and obesity, maintaining disproportion of the limbs and short stature. Based on a suspected diagnosis, by 14 months of age the patient undergone several laboratory examinations.
Pseudo-hypoparathyroidism, first described in 1942, was the first human disorder of hormone resistance to be identified. It is a genetically heterogeneous condition characterized by hypocalcemia and hyperphosphatemia resulting from end-organ resistance to PTH, which is generally elevated. The hormone resistance is usually caused by defects on the ɑ-subunit of the stimulatory form of the GTP-binding protein (Gsɑ), which is a downstream signaling protein of the PTH receptor and of other G protein-coupled hormone receptors, including thyroid-stimulating hormone (TSH), gonadotropins, and growth hormone-releasing hormone (GHRH), which is why patients with PHP may also show resistance to other hormones, such as TSH. Gsɑ is encoded by the GNAS gene, located on chromosome 20q13.3, a complex imprinted locus that also produces additional coding and noncoding transcripts through the use of alternative promoters and alternative splicing, in a tissue-specific manner. Inheritance is autosomal dominant with parental imprinting. Several variants of PHP are recognized on the basis of clinical, biochemical, and genetic features (Table 1).
Diagnosis: Pseudohypoparathyroidism 1a
Parathyroid hormone (PTH) was 329.20pg/mL (reference 15.0-65.0pg/mL), serum calcium 10.3mg/dL (reference 8.4-10.2mg/dL) and phosphorus 7.1mg/dL (reference 2.7-4.5mg/dL), raising the possibility of pseudohypoparathyroidism 1a (PHP-1a) and prompting the genetic testing. The diagnosis was confirmed with the identification of an inactivating mutation of the GNAS gene on the 20 chromosome (c.1096G>A). Parents had normal PTH, calcium and phosphorus levels, however the mother had a very similar phenotype, probably due to a pseudo-pseudohypoparathyroidism.
Discussion
| PHP-Ia | PPHP | PHP-Ib | PHP-Ic | PHP-II | |
|---|---|---|---|---|---|
| AHO manifestations | Yes | Yes | No | Yes | No |
| Serum calcium | ↓ | Normal | ↓ | ↓ | ↓ |
| Serum Phosphate | ↑ | Normal | ↑ | ↑ | ↑ |
| Serum PTH | ↑ | Normal | ↑ | ↑ | ↑ |
| Other hormonal resistance | Yes | No | No, except for some cases | Yes | No |
| Heterotopic ossification | Yes (superficial) | Yes (superficial) | No | Yes (superficial) | No |
| Urinary cAMP after PTH stimulation | ↓ | ↑ | ↓ | ↓ | ↑ |
| Urinary phosphate after PTH stimulation | ↓ | ↑ | ↓ | ↓ | ↓ |
| Inheritance | AD | AD | AD or sporadic | AD | Sporadic |
| GNAS defect | Maternal inactivating mutations | Parental inactivating mutations | Imprinting defects | Maternal inactivating mutations (rare) | Sporadic or none |
Table 1: Clinical, biochemical, and genetic features of Pseudo-hypoparathyroidism.
Table 1: Clinical, biochemical, and genetic features of Pseudo-hypoparathyroidism. AD - autosomal dominant; AHO – Albright Hereditary Osteodystrofia; PHP – pseudo-hypothyroidism; PPHP – pseudo pseudo-hypoparathyroidism; PTH – parathyroid hormone; TSH - Thyroid Stimulating hormone_. (_Adapted from Lemos MC, Thakker RV. GNAS Mutations in pseudohypoparathyroidism type 1a and related disorders [1].
Although rare, PHP-1a is the most frequent variant, with an estimated incidence of approximately 0.79 per 100 000, and corresponding to 50% of cases.
Clinical Features
PHP-1a is highly heterogeneous, and deeply impairing disorder. Patients with PHP-1a have a typical phenotype, described for the first time by Albright, and therefore called Albright’s Hereditary Osteodistrophy (AHO). It is characterized, as in the clinical case presented and evident in Figure 1, by a round face, flat, wide and low nasal bridge, short neck, obesity, short stature, subcutaneous calcifications and skeletal abnormalities (genu valgum, cubitus valgus, brachydactyly of the 4th and 5th fingers).
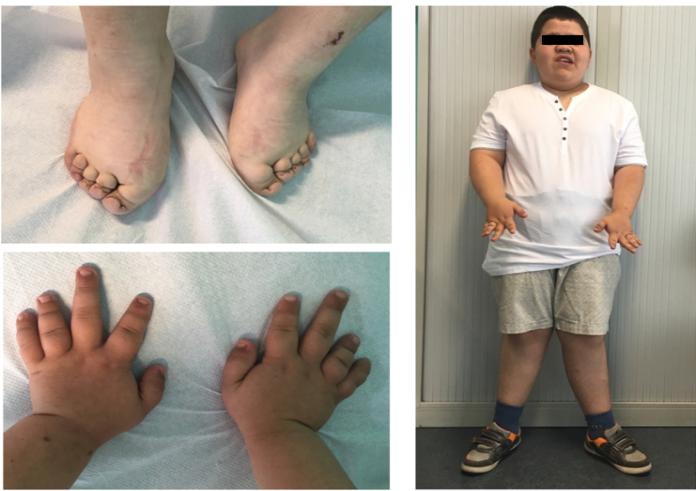
Figure 1: Clinical features of PHP-1a. Brachydactyly (i.e. shortening of the third, forth and/or fifth metacarpal bones, and the first distal phalanx), as well as the subcutaneous calcifications, are the most specific characteristics of AHO. Mental retardation is seen in 50-75% of patients, although frequency and severity of this sign is not well established. Dental dysplasia is also frequent, including aplasia, thin enamel with enlarged pulp chamber, hypoplasia, hypodontia, pulp calcification, multiple carious teeth, multiple unerupted teeth, crowded anterior teeth, anterior open bite, gingival hyperplasia, gingivitis with spontaneous bleeding and pain.
Usually, the phenotype manifests itself in the first year of life, but is usually progressive, with accentuation of the morphological features, and can only be evident after the age of 4, often responsible for a delay in the diagnosis. For unknown reasons, biochemical changes of PHP rarely occur in the first years of life, but hypothyroidism usually precedes PHP and may occasionally be congenital, as in the clinical case presented. Hypocalcemia, rarely occurring before the age of 3, may lead to neuromuscular symptoms, including seizures. Some patients, however, remain normocalcemic. Pubertal delay, amenorrhea or oligomenorrhea, infertility, as a sign of resistance to gonadotropins, may occur. Resistance to GHRH is not universal and so growth rate should be monitored and a GH deficit excluded in order to initiate treatment in a timely manner. Lenticular cataracts appear in patients with a long disease course without appropriate treatment. Other ophthalmologic manifestations include nystagmus, microphthalmia, strabismus, optic nerve atrophy. Patients with pseudo-pseudohypoparathyroidism (PPTH) exhibit most of the somatic features of AHO in the absence of PTH resistance. Both PPHP and PHT-1a result from inherited inactivating mutations on GNAS gene and can coexist in the same family, but never on the same sibship. The development of one disorder or the other depends on the gender of the parent transmitting the genetic defect, as the hormone resistance is parentally imprinted. Thus PTH-1a occur in a child only when the mutation is inherited from the affected mother (as in this case); and PPHP occurs when the mutation is inherited from the affected father.
Radiological Findings
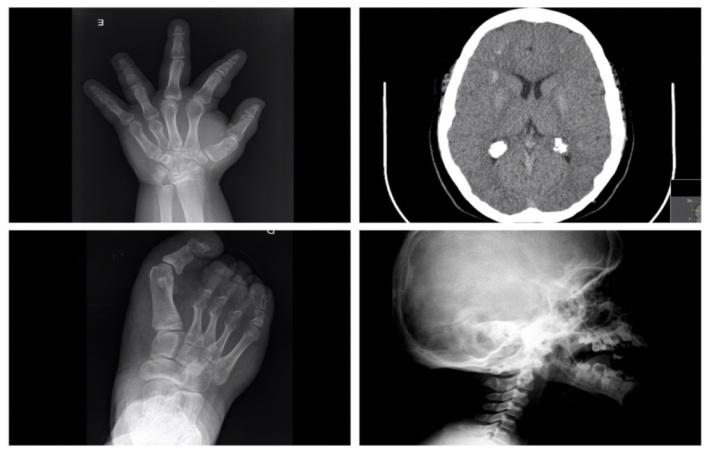
There are generalized changes in osteogenesis with osteoporosis and a delayed bone age versus chronological age. Frontal hyperostosis, craniosynostosis, dolichocephaly and cranial asymmetries may occur. The effect of PTH resistance on the bone is not conclusive, with variable described manifestations, ranging from decreased bone density to over osteitis fibrosa cystica to osteosclerosis. Hand x-ray can show shortening of the metacarpals of the 3rd, 4th and/or 5th finger, as well as of the distal phalange of the 1st finger (Figure 2), with or without heterotopic calcifications. Basal ganglia and frontal subcortical substance calcifications are very typically seen.
Treatment
Treatment of PHP is similar to hypoparathyroidism, with oral calcium and calcitriol. Calcitriol allows to overcome the enzymatic blockade and, when associated with calcium, improves hypocalcaemia. Reaching normal levels of PTH should be tried, as much as possible, to avoid the demineralizing effect. As there seems to be no risk of hypercalciúria, as opposed to patients with hypoparathyroidism, treatment with oral calcium may be more permissive.
Patient Course
The patient kept normal calcium levels till the age of 4, when he presented values of 7.7mg/dL (reference 8.6- 10.2mg/dL) and started calcium and vitamin D supplementation. He maintained hypocalcemia very difficult to normalized, although asymptomatic. Because of recurrent intense headache, with multiple emergency department visits, he underwent computer tomography that revealed typical images of basal ganglia calcification (Figure 2B). Currently with 15 years old, he has typical Albright Hereditary Osteodystrophy phenotype (Figure 1).
Summary
- PHP-Ia is a rare and profoundly disabling condition.
- Diagnosis is not always easy since the phenotypic features may not be noticeable at birth and in the first years of life, and may be heterogeneous later on.
- Endocrine and metabolic changes, although mandatory for the investigation, may not be present or emerging at different times and with varying severity.
- A high degree of suspicion is necessary.
References
-
Lemos MC, Thakker RV (2015) GNAS mutations in Pseudohypoparathyroidsm type 1a and related disorders. Hum Mutat 36(1): 11-19.
-
Mantovani G (2011) Pseudohypoparathyroidism: Diagnosis and treatment. J Clin Endocrinol Metab 96(10): 3020-3030.
-
Sanz-Fernández M, Muñoz-Calvo MT, Pozo-Román J, Martos-Moreno GA, Argente J (2015) Clinical- radiological aspects in a case of pseudohypoparathyroidism type 1a. Hereditary osteodystrophy of Albright. Annals of Pediatrics 82(6): 439-441.
-
Oliveira MR, Bandeira AO, Rendeiro P, Borges TS, Cardoso H (2010) Seudohipoparathyroidism type Ia. An original mutation. Annals of Pediatrics 72(6): 424- 427.
-
Stieler K, Schnabel D, Atugoda S, Sterry W, Blume- Peytavi U (2011) Albright hereditary osteodystrophy. Pediatr Dermatol 28(2): 135-137.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty