Density Functional Study of Aflatoxin B1, B2, G1 and G2: NBO and AIM Analyses
Aflatoxin, belonging to the bisfuranocoumarine group of organic compounds, encompasses several species such as aflatoxin B1, B2, G1, and G2. In this study, geometry optimizations of these aflatoxins were conducted using density functional theory at the B3LYP/6-311G (d, p) level of theory. The computational findings reveal that aflatoxin B1, known for its toxicity, exhibits greater instability. Atom in molecule analysis reveals the presence of a partial covalent intra-molecular bond at C6- O5. Furthermore, the natural bond orbital calculations demonstrate that the structures of the highest occupied molecular orbital and the lowest unoccupied molecular orbital are identical for all four studied aflatoxins, albeit with different energies. Aflatoxins B1 and B2 are determined to be harder species compared to aflatoxins G1 and G2.
H Mohammadi-Manesh1*, F Pourroustaei-Ardakanib2 and SR Fani3
Keywords: Aflatoxin; Atom in Molecule Theory; Density Functional Theory; Natural Bond Orbital
Introduction
Mycotoxins, which are secondary metabolites, produced by fungi such as Penicillium, Fusarium, and Aspergillus, are among the most common natural contaminants in food. These toxins have the potential to infect agricultural crops, posing a significant risk to the food supply. Aflatoxins, famoxides, doxinivalenol, ochratoxin, and zearelenone are examples of the various types of mycotoxins that exist. These mycotoxins pose a significant threat to the health of both humans and animals. Importantly, mycotoxins have a higher carcinogenic risk compared to other known carcinogens [1, 2].
Aflatoxins belong to the group of polyketide mycotoxins and are produced by several species within the Aspergillus genus. A. flavus spores naturally occur in the air, soil, and environment, and can contaminate food products through fungal growth. The most significant and biologically active aflatoxins studied in this paper are aflatoxin B1 (AFB1), aflatoxin B2 (AFB2), aflatoxin G1 (AFG1), and aflatoxin G2 (AFG2) [3, 4]. Figure 1 illustrates the molecular structures of these aflatoxins. AFB (1, 2) molecules contain six oxygen atoms, while AFG (1, 2) molecules contain seven oxygen atoms. The two carbonyl oxygen atoms in these structures play a crucial role in interactions with other molecules. AFB1 and AFG1 have a single bond between the carbon atoms, whereas AFB2 and AFG2 possess a double bond, as depicted in Figure 2.
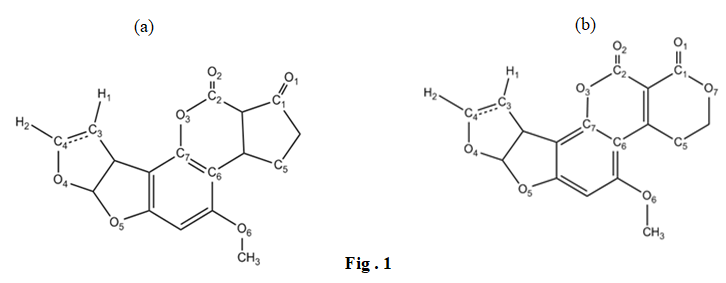
Due to their low molecular weight, aflatoxins in their unchanged state do not typically induce an immune response. Aflatoxins B and G were initially discovered and isolated by a group of researchers in England and the Netherlands. Aflatoxin B1 (C17H12O6) and G1 (C17H12O7) are two commonly studied forms of aflatoxins. Aflatoxins B2 and G2 are derivatives of B1 and G1, respectively, and share structural similarities with their parent compounds. As a result, their spectral properties, such as ultraviolet and infrared spectra, exhibit close resemblance to each other [5, 6].
In recent years, computational methods have been increasingly employed to investigate the characteristics of aflatoxin B1. Nicolás-Vázquez and colleagues utilized quantum mechanics calculations to elucidate the chemical behavior of the lactone ring in aflatoxin B1. Their calculations demonstrated that the lactone ring undergoes hydrolysis in acidic conditions through two molecular mechanisms, thereby diminishing the carcinogenic properties of aflatoxin B1 [7].
Molecular dynamics simulations were conducted to investigate the binding mechanism between aflatoxin B1 and smectite, providing insights into their interaction [8]. Karabulut, et al. focused on the selective reduction of aflatoxin B1 carbonyls and examined the molecular structure of aflatoxicol and its impact on toxicity using density functional theory (DFT) [9]. Their calculations revealed that among the three possible tautomers of aflatoxicol, only one tautomer remained stable in both gas and solution phases. The electronic properties of aflatoxicol were found to be similar to those of aflatoxin B1, which may explain the comparable carcinogenicity and toxicity of these compounds, as supported by experimental results.
This study aimed to investigate the structural properties of aflatoxins B1, B2, G1, and G2, as previous research has predominantly focused on aflatoxin B1 with fewer computational studies conducted on aflatoxin B2, G1, and G2. To achieve this objective, various methods including density functional theory, natural bonding orbital (NBO) program, and the theory of atom in molecule (AIM) were employed. These methods were utilized to calculate the electronic and thermodynamic properties of the aflatoxins and analyze their intramolecular bonds. By employing these computational approaches, a comprehensive understanding of the structural characteristics of aflatoxins B1, B2, G1, and G2 was achieved, shedding light on their unique properties and potential interactions.
Computational Details
To achieve the optimal structure of the aflatoxin composition, it is crucial to obtain the most stable structure with a global minimum. While Monte Carlo methods and molecular dynamics are commonly employed to search for global minima, there is no guarantee that these methods will always lead to the global minimum. Therefore, it is advantageous to utilize various algorithms and pre- optimization techniques with other software [10, 11, 12].
The initial structure of the aflatoxins under investigation was obtained from x-ray crystallography data. Subsequently, the most stable conformers were optimized using the B3LYP (Becke–Lee–Yang–Parr) version of DFT method with the 6-311G (d, p) basis set. All calculations were performed using the Gaussian 03 program [13]. The chosen basis set contains an appropriate number of basis set functions to accurately reproduce experimental data [14, 15]. The absence of negative frequencies in the normal mode analysis confirms that the optimized structures correspond to real minimum stationary points on the potential energy surfaces.
Visualizations of the optimized structures were generated using Gauss View 5.0 [16].
For further analysis, the Natural Bond Orbital (NBO) version 3.1 [17] was employed to perform NBO analysis. Additionally, the AIM2000 software [18, 19] was utilized for Quantum Theory of Atoms in Molecules (QTAIM) analysis. These analyses provide valuable insights into the electronic and intramolecular bonding properties of the studied aflatoxins.
Results and Discussions
Energies and Geometries
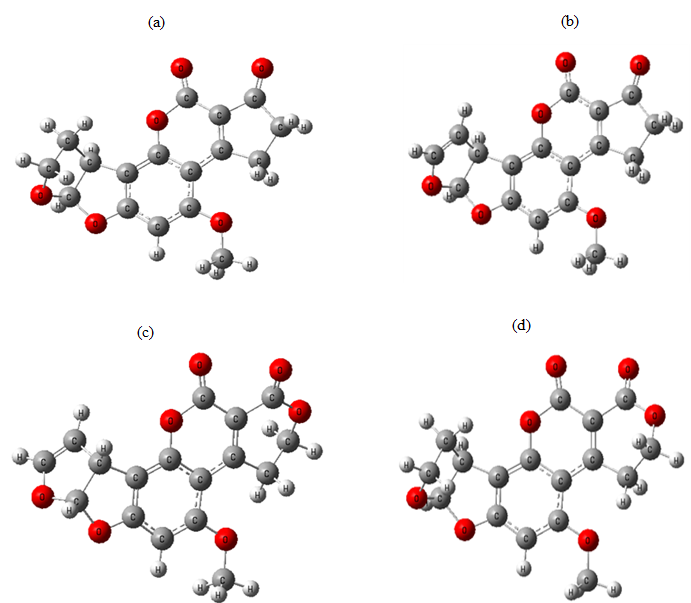
The optimization of the aflatoxin structures provides valuable information regarding their three-dimensional arrangement. Figure 2 illustrates that the optimized structures of the studied aflatoxins are non-planar, and the orientation of the pentagon ring varies among them.
Table 1 presents the distances between selected atoms and the bond lengths of specific bonds in the aflatoxin molecules. It is evident that in AFB (1, 2), the distance between the oxygen atoms of the carbonyl groups (O1 and O2) is greater compared to AFG (1, 2). Additionally, the bond length between O1 and C1 in AFB (1, 2) is longer than that in AFG (1, 2). This suggests that O1 is more strongly bound to its pentagonal ring, allowing the two oxygen atoms (O1, O2) to be more spatially separated and interact with other molecules more effectively. On the other hand, the bond length between O2 and C2 is less influenced by the adjacent ring.
Furthermore, there is a possibility of forming an intermolecular bond between the O6 and C5 atoms, which will be further discussed in the AIM analysis. Interestingly, the distance between O6 and C5 in AFB (1, 2) is greater than in AFG (1, 2), indicating a weaker intermolecular bond between these atoms in AFB (1, 2).
| Species | O -O 1 2 | O -C 1 1 | O -C 2 2 | C -C 3 4 | O -C 6 5 |
|---|---|---|---|---|---|
| AFB1 | 3.0919 | 1.2069 | 1.1929 | 1.3271 | 2.8029 |
| AFB2 | 3.0929 | 1.2071 | 1.1931 | 1.5278 | 2.8018 |
| AFG1 | 2.8351 | 1.1984 | 1.1929 | 1.3271 | 2.7461 |
| AFG2 | 2.8349 | 1.1985 | 1.1932 | 1.5278 | 2.7449 |
Table 1: Atomic distances and bond lengths (in _Å_ ) of understudied aflatoxins at the B3LYP/6-311G (d, p) level of theory in the
Thermodynamic Properties
The thermodynamic properties of the studied aflatoxins were computed using the B3LYP/6-311G (d, p) level of theory at the standard conditions of 298.15 K and 1 atmospheric pressure. The calculated thermodynamic properties are summarized in Table 2. Based on the reported results, it is observed that aflatoxin B1, which is known to be more toxic according to previous studies [3], exhibits higher instability compared to the other aflatoxins. Additionally, it is noted that the dipole moment of AFB2 is weaker than that of the other aflatoxins.
| Species | E ZPE (a.u*) ele | G(a.u.) | H(a.u.) | S(cal.mol-1.K-1) | μ(Debye) |
|---|---|---|---|---|---|
| AFB1 | -1106.3655 | -1106.4121 | -1106.3466 | 138.059 | 8.2512 |
| AFB2 | -1107.5750 | -1107.6221 | -1107.5558 | 139.596 | 7.8513 |
| AFG1 | -1181.5997 | -1181.6470 | -1181.5802 | 140.748 | 9.2389 |
| AFG2 | -1182.8092 | -1182.8570 | -1182.7894 | 142.452 | 8.9194 |
Table 2: Corrected electron energy values and thermodynamic properties of the studied species at B3LYP/6-311G (d, p) level of the
*Atomic unit: 1 au = 627.5095 Kcal. mol−1 Table 2: Corrected electron energy values and thermodynamic properties of the studied species at B3LYP/6-311G (d, p) level of theory in the gas phase.
Natural Bond Orbital (NBO) Analysis
The NBO analysis is a method that involves converting the wave function into its constituent parts. This analysis allows for the determination of natural atomic orbitals, natural hybrid orbitals, natural bond orbitals, and natural local molecular orbitals, which are then used for population analysis and NBO energy analysis. Natural bond orbitals are special vectors derived from the density matrix, formed by the first and second blocks. The central blocks describe internal and nonbonding orbitals, while the two-block consists of bonding and nonbonding orbitals [20, 21, 22].
Table 3 presents the electronic properties of the studied aflatoxins, including the lowest unoccupied molecular orbital (LUMO) and its energy (ε LUMO), as well as its occupation number. It also includes the highest occupied molecular orbital (HOMO) and its energy (ε HOMO), along with its occupation number. Other properties such as ionization energy (I), electron affinity (A), chemical hardness( ) η , electronic chemical potential( ) µ , electrophilicity index( ) ω , and softness (S) are also provided.
The electrophilicity index( ) ω , introduced by Parr, et al. [23, 24, 25, 26], and is a descriptor used to quantify the global electrophilic nature of a molecule on a relative scale. Chemical hardness, electronic chemical potential, and softness are universal reactivity descriptors [27, 28]. The stability of a chemical species can be related to its hardness, where soft molecules have a small energy band gap between the LUMO and HOMO, while hard molecules have a larger gap [29, 30].
Based on the results in Table 3, it can be observed that despite the structural and reactivity differences among the aflatoxins, their HOMO and LUMO orbitals have the same characteristics but with different energy levels. Additionally, the band gap for AFG1 and AFG2 (6.5) is smaller than that of AFB1 and AFB2 (6.6). This suggests that AFB1 and AFB2 are harder species compared to AFG1 and AFG2, as indicated by the higher energy band gap.
| Electronic Properties | Formula | AFB1 | AFB2 | AFG1 | AFG2 |
|---|---|---|---|---|---|
| Lowest Unoccupied Molecular Orbital | LUMO | BD1 C -C *(2) 6 7 | BD* C -C (2) 6 7 | BD* C -C (2) 6 7 | BD* C -C (2) 6 7 |
| Occupation Number of LUMO | - | 0.4789 | 0.4803 | 0.4766 | 0.4785 |
| Energy of LUMO (eV) | εLUMO | -0.0707 | -0.068 | -0.1551 | -0.1333 |
| Highest Occupied Molecular Orbital | HOMO | LP O (2) 1 | LP O (2) 1 | LP O (2) 1 | LP O (2) 1 |
| Occupation Number of HOMO | - | 1.8611 | 1.8655 | 1.8309 | 1.8311 |
| Energy of HOMO (eV) | εHOMO | -6.7022 | -6.6968 | -6.6478 | -6.626 |
| Band gap (eV) | ∆E (L −H) | 6.6315 | 6.6288 | 6.4927 | 6.4927 |
| Ionization Energy(eV) | I = −(εHOMO) | 6.7022 | 6.6968 | 6.6478 | 6.626 |
| Electron Affinity (eV) | A= −(εLUMO) | 0.0707 | 0.068 | 0.1551 | 0.1333 |
| Chemical Hardness (eV) | (I−A) η= 2 | 6.6668 | 6.6628 | 6.5702 | 6.5593 |
| Electronic Chemical Potential (eV) | −(1+A) η= 2 | -3.3864 | -3.3824 | -3.4014 | -3.3796 |
| Electrophilicity Index (eV) | µ2 ω= η | 1.7202 | 1.7171 | 1.7609 | 1.7413 |
| Softness (1/eV) | 1 S = η | 0.1499 | 0.1501 | 0.1522 | 0.1525 |
| Atom | AFB1 | AFB2 | AFG1 | AFG2 | |
| O 1 | -0.5328 | -0.5342 | -0.5349 | -0.5357 | |
| O 2 | -0.5241 | -0.5232 | -0.5227 | -0.5239 | |
| O 3 | -0.5251 | -0.5318 | -0.5238 | -0.5261 | |
| O 4 | -0.5411 | -0.5669 | -0.5417 | -0.5664 | |
| O 5 | -0.5268 | -0.5355 | -0.5275 | -0.5352 | |
| O 6 | -0.5217 | -0.5234 | -0.5297 | -0.5302 | |
| O 7 | - | - | -0.5475 | -0.5479 |
Table 3: Calculated electronic properties for the studied species at the B3LYP/6-311G (d, p) level of theory in the gas phase.
BD*, 2-Center Antibonding Orbital LP is Valence Lone Pair Table 3: Calculated electronic properties for the studied species at the B3LYP/6-311G (d, p) level of theory in the gas phase.
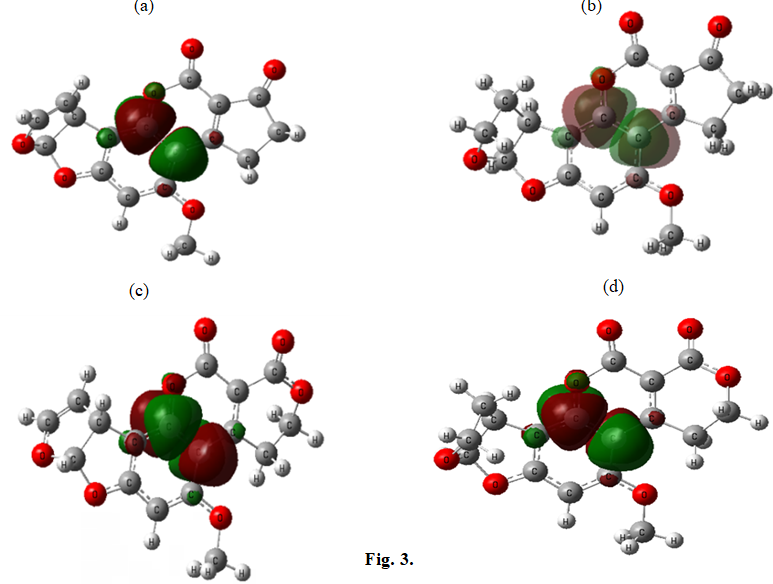
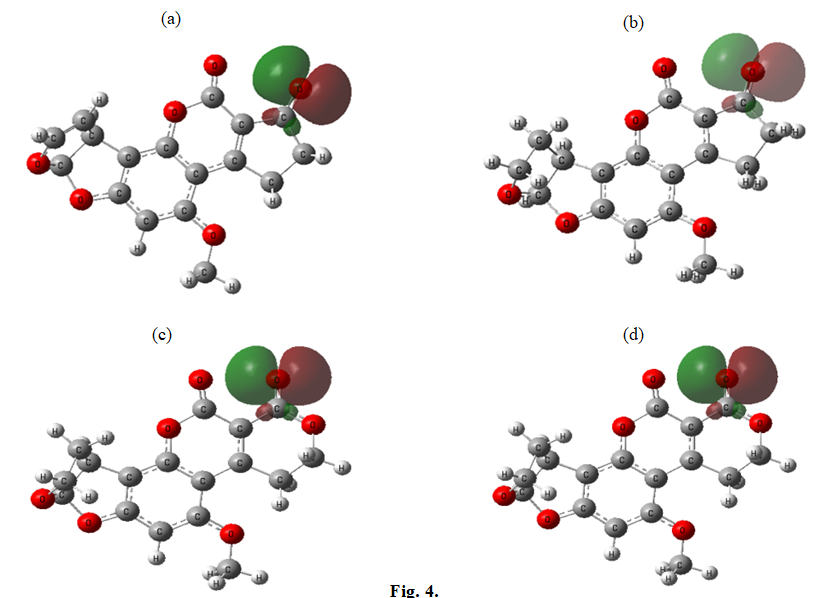
Figures 3 & 4 depict the HOMO and LUMO orbitals of the studied aflatoxins, illustrating that the HOMO and LUMO orbitals are the same across all aflatoxins.
Table 4 presents the natural charges of the important atoms in aflatoxins B1, B2, G1 and G2, obtained using the B3LYP/6-311G (d, p) level of theory. From Table 4, it can be observed that the partial charge of O1 is greater than that of O2. Additionally, as indicated in Table 3, the HOMO orbital is primarily located on the O1 atom. Therefore, the O1 atom exhibits a higher tendency to react with other molecules based on its higher partial charge and its involvement in the HOMO orbital.
Atoms in Molecules (AIM) Analysis
In AIM theory, critical points (CPs) are points where the gradient of the electron density becomes zero. There are four categories of critical points, which are determined by the values of the hessian matrix of the electron density. The hessian matrix contains the second derivatives of the electron density with respect to various coordinate axes, and its eigenvalues and eigenvectors are denoted as follows:
Here, i
λ represents the eigenvalues and ui represents
the eigenvectors for the ith critical point. Each critical point
is characterized by its degree and symbol (r, s), where r is
the number of non-zero eigenvalues and s is the sum of the
algebraic signs of the eigenvalues. For example, a bond critical
point (BCP) typically exists between adjacent nuclei and is
represented as (1, 3). This critical point has two negative
$$ \lambda 1 \leq \lambda 2 \leq 0 a $$
and one positive eigenvalue 3
0 λ >
. Table 5 provides a breakdown of critical points based on the
values of r and s.
The trajectory of electron density starting from a BCP and ending at each nucleus is referred to as the pathway. The molecular graph consists of a network of bond paths, which are closely related to the chemical bonding network in the molecule’s equilibrium geometry. The electron density along the bond path at the BCP reaches its lowest value, indicating a strong chemical bond.
The electron density ( ) r ρ is often used to describe the strength of chemical bonds. Higher values of ( ) r ρ correspond to stronger bonding. The Laplacian of the electron density $$ \left(\nabla^ {2} \rho (r)\right) $$
is the sum of the second derivatives of the
electron density along the coordinate axes at a given point
r. The Laplacian values provide information about bond
characteristics. A negative value of
( ) ² r ρ ∇
and high values
of ( ) r ρ
indicate covalent bonding, while a positive value of
( ) ² r ρ ∇
and low values of
( ) r ρ
are associated with closed-
shell interactions, including ionic, coordinate, hydrogen, and
van der Waals bonds. In systems involving hydrogen bond interactions, rare gas dimers, or ion systems, the electron density is approximately one hundredth of an atomic unit, and the Laplacian is positive.
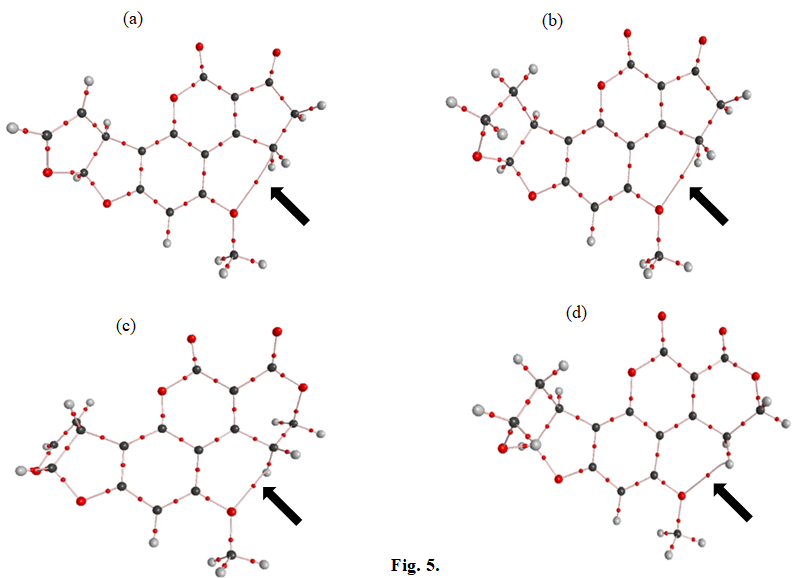
Figure 5 shows the critical points of electron density distribution and the gravitational pathways, where an intermolecular bond between the O6 and C5 atoms is indicated by an arrow. Table 5 summarizes the values of electron density ( ) ( ) r ρ , electron density Laplacian ( ) 2 ( ) r ρ ∇ , total energy ( ) ( ), 2 H r V G ratio, and ellipticity (ε ) at the BCP for the studied aflatoxins.
| Name of Critical Point | Symbol | λ 1 | λ 2 | λ 3 | (r, s) |
|---|---|---|---|---|---|
| Cage Critical Point | CCP | + | + | + | (3, +3) |
| Ring Critical Point | RCP | - | + | + | (3, +1) |
| Bond Critical Point | BCP | - | - | + | (3, -1) |
| Non-Nuclear Point | NNP | - | - | - | (3, -3) |
Table 4: Classification of Critical Points Based on their Types.
In Table 6, the small values of ρ(r) and the positive sign of the Laplacian indicate that the electron density is concentrated in the interaction region between the O6 and C5 nuclei, suggesting a closed-shell interaction for the O6- C5 molecular bond. Furthermore, by comparing the values of ρ(r) for this bond in different aflatoxin types, it can be concluded that the O6-C5 bond is stronger in aflatoxins AFG (1, 2) compared to the others.
The electron energy density, represented by the Hamiltonian, is negative for collective interactions, with the absolute value indicating the dominance of potential energy in the covalent character of the interaction. The quantities
V(r) and G(r) represent potential energy and kinetic energy,
respectively, and the ratio
2 V
G is always positive. If this ratio
is greater than 1, it indicates a negative Laplacian
$$ \left(\nabla^ {2} \rho (r)\right) $$
and a covalent interaction. If the ratio falls between 0.5 and
1, it implies a positive Laplacian and a negative Hamiltonian
(H(r)), indicating a partial covalence interaction. When the
ratio is less than 0.5, both the Laplacian and Hamiltonian are
positive, suggesting a non-covalent interaction. Therefore,
for the O6-C5 interaction, the ratio falls within the range of
0.5-1, indicating a partial covalence bond.
Regarding the C1-O1, C2-O2, and C3-C4 bonds, they exhibit covalent couplings, with the C3-C4 bond being weaker due to its lower ρ(r) value. The ellipticity values (ε ) indicate the proximity of curves perpendicular to the bond path, with a higher ε value suggesting a higher percentage of π-type bond participation. In the case of the O6-C5 interaction, the ε value is greater than for other bonds, indicating a higher degree of π-type bond character and participation [31, 32].
Aflatoxin BCP ( )* r ρ ( )* ² r ρ ∇ ( )* H r ε AFB1
AFB2
AFG1
AFG2
2 V G C1-O1 0.4135 -0.0585 0.6958 0.0541 4.0429 C2-O2 0.4313 -0.1215 0.743 0.1203 4.0853 C3-C4 0.3499 -1.0442 0.4131 0.4254 7.4337 O6-C5 0.0123 0.0549 -0.0021 1.8102 1.6429 C1-O1 0.4123 -0.0672 0.6931 0.0539 4.0497 C2-O2 0.4308 -0.1205 0.7416 0.1194 4.0847 C3-C4 0.2473 -0.5687 0.1998 0.0279 8.9293 O6-C5 0.0125 0.0559 -0.0021 1.9248 1.6441 C1-O1 0.4258 -0.1422 0.7297 0.1133 4.1024 C2-O2 0.4311 -0.1193 0.7429 0.1202 4.0837 C3-C4 0.3479 -1.0311 0.4082 0.4257 7.4262 O6-C5 0.0188 0.0759 -0.0027 0.3701 1.6674 C1-O1 0.4257 -0.1432 0.7295 0.1132 4.1032 C2-O2 0.4309 -0.1213 0.7423 0.1199 4.0852 C3-C4 0.2473 -0.5686 0.1998 0.0279 8.9297 O6-C5 0.0189 0.0763 -0.0027 0.3706 1.6673
| $ | 6 | 5 |
Table 5: Topological Characteristics of Selected Critical Points in the Studied Aflatoxins.
Conclusions
This study provided insights into the structural properties of aflatoxin B1, B2, G1, and G2 using computational methods. The results indicated that aflatoxin B1, known for its toxicity, is less stable compared to the other aflatoxins. The O1 atom in aflatoxin B1 and B2 exhibited higher reactivity and attachment to the pentagonal ring, along with a greater charge. This suggests that it has a stronger interaction with other substances.
The bond length between O1 and C1 atoms was found to be longer in aflatoxin B1 and B2. The AIM analysis revealed the presence of an intermolecular bond between C5 and O6, indicating a partial covalence bond. The natural bond orbital analysis showed that the energy and structure of HOMO and LUMO were the same for all four aflatoxins studied. Additionally, the larger energy band gap in aflatoxin B1 and B2 indicated that they are harder species compared to aflatoxin G1 and G2. These findings contribute to a better understanding of the structural and electronic properties of aflatoxins and their reactivity.
Acknowledgments
The authors would like to acknowledge the computational support provided by the Supercomputing Center of Yazd University. Their assistance was instrumental in conducting the computational calculations and analysis for this research. Additionally, the authors extend their gratitude to Dr. Claudia Probst for her valuable comments, which contributed to the improvement of this study.
Conflicts of Interest
The Authors state that there is no conflict of interest.
Funding
There are not any research grants or support for this manuscript. The Authors declare that there are no conflicts of interest regarding the publication of this manuscript. Furthermore, there are no specific research grants or funding sources to disclose for this particular study.
References
-
Amaike S, Keller NP (2011) Aspergillus flavus. Annu Rev Phytopathology 49: 107-133.
-
Wang JS, Groopman JD (1999) DNA damage by mycotoxins. Mutat Res 424(1-2): 167-181.
-
Fan S, Zhang F, Liu S, Yu C, Guan D, et al. (2013) Removal of aflatoxin B(1) in edible plant oils by oscillating treatment with alkaline electrolysed water. Food Chem 141(3): 3118-3123.
-
Fani SR, Moradi M, Probst C, Zamanizadeh HR, Mirabolfathy M, et al. (2014) A critical evaluation of cultural methods for the identification of atoxigenic Aspergillus flavus isolates for aflatoxin mitigation in pistachio orchards of Iran. Eur J Plant Pathol 140: 631- 642.
-
Kalinichenko AA, Toporova VA, Panina A, Aliev T, Kryukova E, et al. (2010) Development and characterization of antibodies against aflatoxins. Russ J Bioorg Chem 36: 114-123.
-
Turner PC, Moore SE, Hall AJ, Prentice AM, Wild CP (2003) Modification of immune function through exposure to dietary aflatoxin in Gambian children. Environ Health Perspect 111(2): 217-220.
-
Nicolás V, Méndez AA, Moreno ME, Miranda R, Castro M (2010) Role of lactone ring in structural, electronic, and reactivity properties of aflatoxin B1: a theoretical study. Arch Environ Contam Toxicol 59(3): 393-406.
-
Deng Y, Szczerba M (2011) Computational evaluation of bonding between aflatoxin B1 and smectite. Appl Clay Sci 54(1): 26-33.
-
Karabulut S, Paytakov G, Leszczynski J (2014) Reduction of aflatoxin B1 to aflatoxicol: a comprehensive DFT study provides clues to its toxicity. J Sci Food Agric 94(15): 3134-3140.
-
Young D, (2004) Computational chemistry: a practical guide for applying techniques to real world problems. General Chemistry John Wiley & Sons.
-
Becke D (1993) Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7): 5648- 5652.
-
Lee, Yang W, Parr RG (1988) Development of the Colle- Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2): 785-789.
-
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2004) Gaussian 03 Revision D.01. Gaussian Inc Wallingford CT.
-
McLean AD, Chandler G (1980) Contracted Gaussian basis sets for molecular calculations. I. Second row atoms. J Chem Phys 72: 5639-5648.
-
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self‐ consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72: 650-654.
-
Dennington R, Keith T, Millam J (2009) GaussView 5.0.8. Shawnee Mission.
-
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1998) NBO 3.1. TCI. University of Wisconsin Madison.
-
Biegler K, Schönbohm J (2002) Update of the AIM2000- Program for atoms in molecules. J Comput Chem 23(15): 1489-1494.
-
Bader RF (1985) Atoms in molecules. Acc Chem Res 18(1): 9-15.
-
Glendening ED, Landis CR, Weinhold F (2011) Natural bond orbital methods. Natural bond orbital methods Comput Mol Sci 2(1): 1-42.
-
Koshland DE (1958) Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci USA 44(2): 211-216.
-
Popelier PLA, Aicken FM, O’Brien SE (2000) Atoms in molecules. The Royal Society of Chemistry 3: 143-198.
-
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity Index. J Am Chem Soc 121(9): 1922-1924.
-
Ajeel FN, Mohammed MH, Khudhair AM (2019) SWCNT as a Model Nanosensor for Associated Petroleum Gas Molecules: Via DFT/B3LYP Investigations. Russ J Phys Chem B 13: 196-204.
-
Fotooh FK, Atashparvar M (2019) Theoretical Study of the Effect of Simultaneous Doping with Silicon, on Structure and Electronic Properties of Adamantane. Russ J Phys Chem B 13(1): 1-8.
-
Pearson RG (1997) Chemical Hardness: Applications from Molecules to Solids. John Wiley & Sons.
-
Parr RG, Zhou Z (1993) Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc Chem Res 26(5): 256-258.
-
Pearson RG (1987) Recent advances in the concept of hard and soft acids and bases. J Chem Educ 64(7): 561- 567.
-
Pearson RG (1988) Absolute electronegativity and hardness: application to inorganic chemistry. Inorg Chem 27(4): 734-740.
-
Bader RFW (1998) A Bond Path: A Universal Indicator of Bonded Interactions. J Phys Chem A 102(37): 7314- 7323.
-
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91(5): 893-928.
-
Matta CF, Boyd RJ (2007) The Quantum Theory of Atoms in Molecules. John Wiley & Sons.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State