Development of an Antihypertensive Loaded Solid Lipid Nanoparticle
Solid lipid nanoparticles were prepared by using the solvent evaporation method. The prepared solid lipid nanoparticles of felodipine from different batches were weighed accurately and filled into appropriate-sized capsules for further evaluation. The prepared nanoformulation was tested in vitro for drug entrapment efficiency found to be 97-99% for the formulation with soya lecithin and glyceryl mono-stearate and percentage yield. The results of the in vitro drug release study for all formulations performed at gastric pH 1.2 indicated that the drug was released in an immediate manner within 60 minutes, which could be due to a burst effect or a faster rate of drug release due to smaller particle size SLN13 the concentration of Glycerol Monosterate- 576.522mg and Poloxamer 188. In vitro release data from standardised nanoformulations was fitted to various release kinetics models for the drug release mechanism. The optimised formulation demonstrated improved drug release and entrapment efficiency, and in vitro release data were fitted to various kinetic models to better understand the drug release mechanism. In vitro assay results were ranged between 76.5 to 96%, Zeta potential -40.75 ± 2.33 (mV) ± SD, entrapment efficiency 99.32. Accelerated stability studies for standardised nanoformulations were performed for 6 months in accordance with standard ICH guidelines, and no appreciable changes in the physical appearance, drug content, and in vitro drug release rates of the nanoformulations were observed. As a result of the study's evidence, it is concluded that felodipineloaded solid lipid nanoparticles are the best choice for hypertension.
Introduction
Drug delivery systems are essential for effectively delivering medication molecules to the intended location. The oral route is preferred for long history, ease of use, and patient compliance. It’s extensively investigated in both conventional and novel drug delivery techniques [1]. The oral route of drug administration has been explored as a field of interest in both conventional and novel drug delivery. This route has historically been the main drug delivery mechanism, accounting for patient compliance, ease of administration and cost effective [2, 3].
According to the WHO (World Health Organization) study one billion people are facing to hypertension the top risk factor for mortality are high blood pressure (12.7%
fatalities are attributed to it), smoking (8.7%) and Diabetics (5.8%). At least 7.1 million people worldwide succumb to hypertension annually, among them 31% of women and 37% of men. The transition from infectious diseases to chronic non-communicable conditions like hypertension poses a considerable challenge, particularly in poorer and lower middle-income countries (LMICs).
Hypertension is well-established in developed countries. Hypertension and cardiovascular diseases pose an emerging public health concern in LMICs due to populations age becomes more urbanized, life style changes favor sedentary behaviors, lack of physical activity, obesity, rising alcohol consumption and salt intake. Proper medication and lifestyle interventions LMICs struggle to achieve optimal blood pressure (BP) due in part to low adherence to pharmaceutical and non-pharmacological therapy. Health services should focus on lowering the hypertension as it can be affected by both medication and life style based therapies [4].
Patients are given hypertension medication along with medication to reduce the cardiovascular and renal mortality and morbidity. The risk of CVD-related complications is reduced if blood pressure is maintained under 140/90mmHg. The ideal blood pressure in human with hypertension, diabetes and kidney disease is 130/80mmHg. Antihypertensive drugs can reduce the risk of heart failure by more than 50%, myocardial infraction by 20% - 25% and stroke by 35% - 40% [5, 6]. Treatment for hypertension follows the hierarchy of medication therapy to control the blood pressure along with the lifestyle changes if it is not able to control the first line of treatment include the thiazide diuretics either alone or in combination with other class. To control the BP, majority of hypertension patients require two or more medications from different groups. Patient with other complications like diabetics, chronic renal disease, heart failure, ischemic heart disease or recurrent stroke with require the combination of drugs used to treat the multiple conditions. Either alone Patients can typically transition to longer follow-up periods for determining if their blood pressure levels are stable once a suitable degree of blood pressure control is attained [7, 8].
Aqueous colloidal dispersions with solid biodegradable lipids as their matrix make up solid lipid nanoparticles. They were first made available as a carrier system in 1990 as an alternative to conventional colloidal carriers such as emulsions, liposomes, and polymeric micro and nanoparticles. When compared to traditional carriers like cream, tincture, and emulsion, solid lipid nanoparticles (SLN) have a number of advantages over liposomes, including improved physical stability, a lower cost than phospholipids, and ease of scaling up and manufacturing. SLN also combines the advantages of controlled release, in vivo tolerability, and active compound protection.
The solid lipid core matrix of solid lipid nanoparticles can solubilize lipophilic compounds. Surfactants (emulsifiers) aid in the stability of the lipid core. Triglycerides, such as tristearin; diglycerides, such as glycerol bahenate; monoglycerides, such as glycerol monostearate; fatty acids, such as stearic acid; steroids, such as cholesterol; and waxes are all examples of lipids (e.g: cetyl palmitate). To stabilise the lipid dispersion, various emulsifiers with varying charges and molecular weights have been used. It was discovered that combining emulsifiers could more effectively prevent particle agglomeration.
A submicron particulate medication delivery device known as a “solid lipid nanoparticle” has an average diameter between 10 and 1000nm with a solid lipid matrix. The main characteristics of SLNs in relation to parenteral application are good physical stability, preservation of incorporated labile medicines from degradation, and regulated drug release. Drugs that were weakly lipophilic and hydrophilic could have their bioavailability increased by being encapsulated in SLN. Solid lipid nanoparticles have been created as possible medication delivery systems. The most effective methods for increasing the bioavailability of class II compounds— drug molecules that are poorly water-soluble but highly permeable—have been lipid-based systems [9, 10, 11].
Materials and Methods
Materials
Felodine (Yarrow chem. Product, Mumbai), Chloroform (Rankem RFCL Ltd, New Delhi), Methanol (Rankem RFCL Ltd, New Delhi), Acetone (L.R) (Rankem RFCL Ltd, New Delhi), Soy lecithin (HiMedia Laboratories Pvt. Ltd.), Poloxamer 188 (HiMedia Laboratories Pvt. Ltd.), Glycerol monostearate (Rolex Laboratories Pvt. Ltd.), Potassium di hydrogen phosphate-L.R-(Rankem RFCL Ltd, New Delhi), Sodium hydroxide -L.R (Qualigens Fine chemicals, Mumbai).
Preparation of Felodipine Loaded Solid Lipid Nanoparticles: Solvent evaporation was used to create solid lipid nanoparticles. The lipid was first heated, then melted lecithin added in a beaker, and the medication is mixed in it. The lipid is heated to 50°C Over the melting point to melt the lecithin. Poloxamer 188 was simultaneously dissolved in water and heated to the same temperature as the lipid phase in another beaker. This aqueous phase was then transferred to the lipid phase. This mixture is immediately put in a probe ultrasonicator at 75% amplitude for 25 minutes after being homogenised at 20,000rpm for 15minutes. The prepared solid lipid nanoparticles of felodipine from different batches were weighed accurately and filled into appropriate-sized capsules for further evaluation [12]. The preliminary trials of felodipine loaded nanoparticles were prepared by using different concentration of glycerol monosterate, soy lecithin and Poloxamer188 which is tabulated in Table 1.
| Ingredients | Formulation code Quantity (mg) | |||||||
|---|---|---|---|---|---|---|---|---|
| SLN 1 | SLN 2 | SLN 3 | SLN 4 | SLN 5 | SLN 6 | SLN 7 | SLN 8 | |
| Felodipine | 400 | 400 | 400 | 400 | 400 | 400 | 400 | 400 |
| Glyceryl monosterate | 150 | 300 | 450 | 600 | 450 | 450 | 450 | 450 |
| Soy lecithin | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 |
| Poloxamer 188 | 3000 | 3000 | 3000 | 3000 | 1500 | 4500 | 6000 | 7500 |
Table 1: Preliminary Trials for Felodipine Loaded Solid Lipid Nanoparticles.
Optimization Studies
Selection of Independent Variables and Dependent Variable: Two formulation variables at two levels were chosen: concentration of glycerol monostatic, soyalecithin, and poloxammer 188. The ranges were fixed based on the preliminary studies. Time taken for the 100% drug release had been taken as the dependent variable and independent variable for glycerol monosterate, with a low concentration of 150mg and a high concentration of 600mg, and for polaxomer 188, with a low concentration of 1500mg and a higher concentration of 7500mg.
Method of Optimization
As the independent variables had been selected as two concentrations of glycerol monosterate and concentration of poloxamer 188, and levels were two, a 23 full factorial design was followed to study the effect of the above-mentioned variables on the drug release [13]. The Design Expert 9.0.3.1 software was used for optimising the variables and tabulating them in Table 2.
| Std | Run | Conc. of glycerol monosterate (mg) | Conc. of Poloxamer (g) | Formulation Code |
|---|---|---|---|---|
| 1 | 1 | 150 | 1.5 | SLN 9 |
| 4 | 2 | 600 | 7.5 | SLN 10 |
| 3 | 3 | 150 | 7.5 | SLN 11 |
| 2 | 4 | 600 | 1.5 | SLN 12 |
The formulations were prepared as per the designed formula, and drug release studies were carried out. The obtained in vitro drug release data were then entered into the software to generate the predicted optimized formula (OPT), and the effect of various formulation variables on the drug release kinetics was investigated. Evaluation of Preliminary Formulations and Optimized Formulation: Assay- A centrifuge tube containing 5ml of methanol and 0.1ml of drug-loaded solid lipid nanoparticles was vortexed for 15minutes. The supernatant was then removed after 20minutes of centrifuging the solution at 6000rpm. Then, added 5ml of methanol into the same centrifuge tube, vortexed, and placed again in the centrifuge for 20min. This supernatant is assayed spectrophotometrically for felodipine at 239nm. Entrapment Efficiency: Centrifugation of samples at 10,000rpm for 10minutes was used to test the effectiveness of entrapment of drug-loaded SLNs. By employing the supernatant of unloaded nanoparticles with basic correction, a UV spectrophotometer operating at 239nm was used to quantify the amount of free drug present in the clear supernatant. The following equation could be used to calculate the entrapment efficiency (EE %) [14].
W W EE X W − =
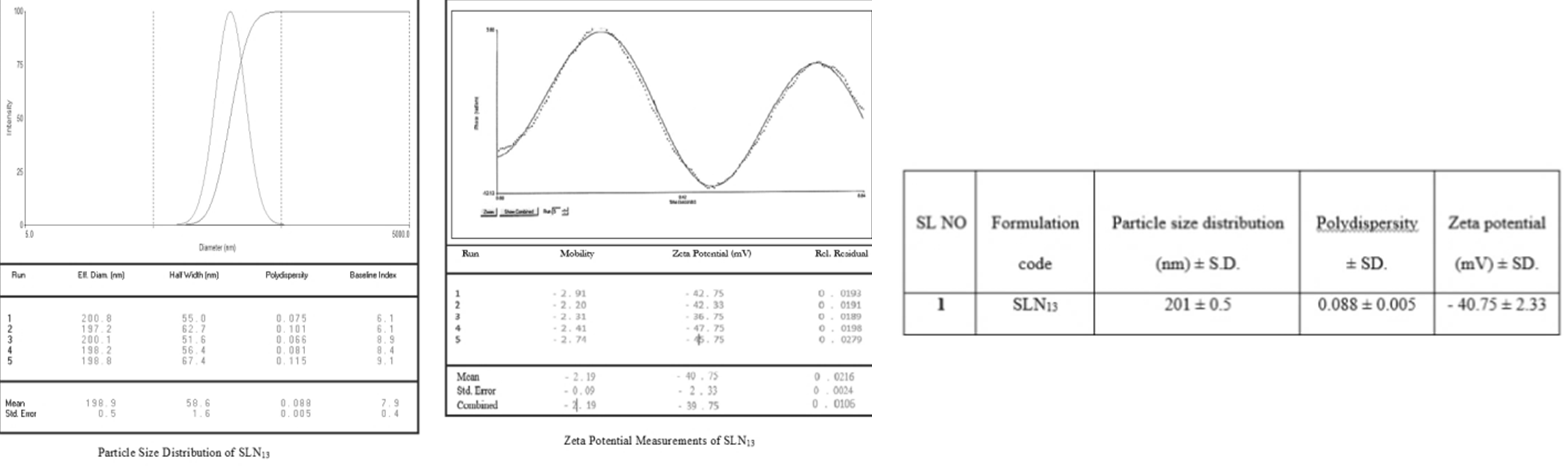
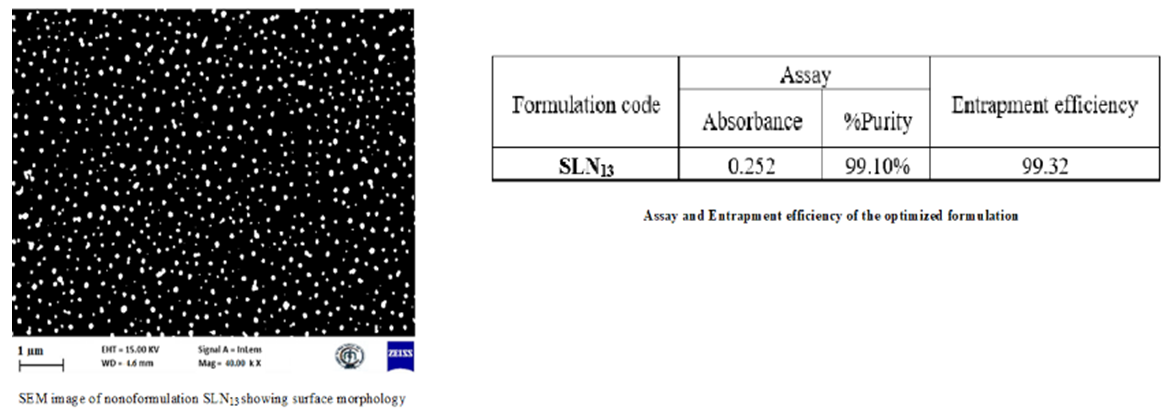
% 100 initial drug free drug initial drug Where “W initial drug” is the mass of the initial drug used and “W free drug” is the mass of the free drug detected in the supernatant after centrifugation of the aqueous dispersion. Particle size of Preliminary formulation Solid Lipid Nanoparticles: The morphology and average particle size of the solid lipid nanoparticles (SLN1-SLN8) were determined using an optical microscope. Determination of Particle size distribution and polydispersity by dynamic light scattering (SLN9-SLN13): The particle size distribution of the solid lipid nanoparticle formulations was measured using dynamic light scattering (DLS) (Brookhaven Instruments, Zetaplas, Germany) by uniformly dispersing the nanoparticles in distilled water at 25°C for a period of 30sec. DLS is based on the principle of “Brownian motion.” Each sample was run for either 3 or 5 runs. Zeta Pals software was used to obtain the particle size distribution graph along with the effective diameter and polydispersity index of the nanoparticles. The polydispersity index refers to how uniformly the nanoparticles are dispersed without aggregation. We also calculated the mean, standard error, combined diameter, and polydispersity for each run. Zeta Potential Measurements (SLN9-SLN13): The zeta potential of the solid lipid nanoparticle was measured to determine the stability of the nano-formulation using a zeta potential analyzer (Brookhaven Instruments, Zetaplas, Germany). Samples were dispersed in distilled water at 250°C for a period of 30sec in a quartz cuvette, with the electrode dipped inside the distilled water in the cuvette. Each sample was run five times, and the Zeta Potential graph was obtained using Zetapals software of Smoluchowski model. Also obtained were the mean, standard error, combined mobility, and zeta potential (mV) of all the runs [15, 16, 17, 18, 19, 20]. Particle Size Measurement by Scanning Electron Microscopy (SLN9-SLN13): Particle’s size Scanning Electron Microscopy (SEM) was used to measure solid lipid nanoparticles (Zeiss Ultra 55, Germany). The nanoparticles were initially adhered to a tiny silicon chip coated in electro conductive paste before being vacuum sputtered with gold. Different magnifications were used to study the nanoparticles’ surface shape and particle size in increasing order.
In-vitro Dissolution Studies: In-vitro release kinetics: Using PCP-Disso - V3 software, the data from in vitro dissolution tests was fitted into the Higuchi matrix, Hixson-Crowell Cube Root Law Model, and Korsmeyer-Pappas equations in order to examine the drug release rate kinetics and mechanism from the formulations of felodipine. Stability Studies: Stability is a desired and required parameter for pharmaceutical products. The purpose of stability studies is to ensure that the drug shows perfect quality throughout its shelf life. A pharmacopoeia product is stable if it is within the limits of its monograph specifications, like its identity, strength, quality, purity etc [21, 22]. The ability of a medicine to be stable has been defined as its ability to remain within its physical, chemical, therapeutic, and toxicological definitions in a certain container and formulation.
Result and Discussion
Analytical Methods
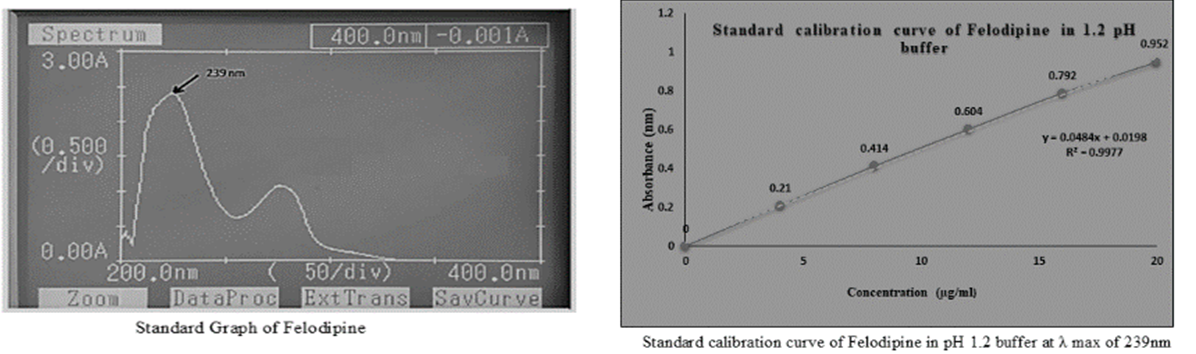
The absorption maxima ( max λ ) were determined using a UV-visible spectrophotometer and a standard calibration curve for felodipine in a pH 1.2 buffer. To determine the wavelength of maximum absorption ( max λ ), the medication solution was scanned in the UV range between 200 and 400nm. The max λ was discovered to be at 239nm, and (Figure 1) depicts the standard calibration curve of felodipine that was developed using 1.2 buffer at the max λ 239nm.
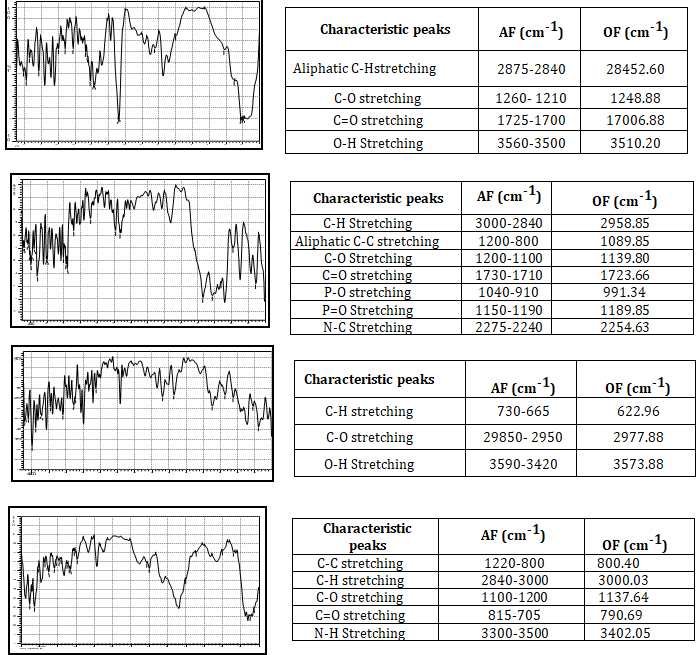
Drug-Polymer Compatibility Studies (Fourier Transfer Infrared Spectroscopy): FT-IR was used to conduct a study on the compatibility of drugs and polymers. The IR spectra of a medication, a polymer, and their physical combination are shown in (Figure 2) in that order. This method involves manually pressing a solid sample into a pellet die after mixing it with KBr, which is transparent to IR, in a mortar and pestle. The samples are then subjected to IR radiation.
Evaluation of Preliminary Formulations: Here, assay, entrapment efficiency, particle count by optical microscopy, and in vitro drug release with a kinetic model are studied, which is shown in (Figure 3). Entrapment efficiency of Felodipine SLNs (SLN1-SLN8) was found to be in the range of 97 to 99 % mentioned in (Table 3) for the formulations prepared with soya lecithin and glyceryl monosterate as lipid materials. Felodipine is essentially water-insoluble and has a high lipophilicity. As a result, during preparation using high-temperature sonication and homogenization, the medication was favorably partitioned towards melting lipids.
| Sl. No | Formulation Code | Assay | Entrapment Efficiency | |
|---|---|---|---|---|
| Absorbance | %Purity | |||
| 1 | SLN 1 | 0.225 | 96% | 98.12 |
| 2 | SLN 2 | 0.211 | 92% | 97.34 |
| 3 | SLN 3 | 0.206 | 88% | 99.18 |
| 4 | SLN 4 | 0.201 | 80% | 99.36 |
| 5 | SLN 5 | 0.221 | 94% | 99.81 |
| 6 | SLN 6 | 0.207 | 84% | 97.45 |
| 7 | SLN 7 | 0.198 | 78% | 99.57 |
| 8 | SLN 8 | 0.194 | 76.5% | 98.90 |
Table 3: Entrapment Efficiency.

The morphology and average particle size of the solid lipid nanoparticles (SLN1–SLN8) were determined using an optical microscope. The particles were found to lie in the size range of 181.34µm to 257.18µm and the particle morphology was also shown in (Figure 3). The resultant particles were found to be almost spherical in shape with a different surface nature. Cumulative drug release for different formulations (SLN1–SLN8) was shown in Figure 3.
A fast release of Felodipine until 60min (62% to 96%) was observed for the formulation prepared with glyceryl monosterate and soy lecithin. In vitro release data obtained from various formulations was fitted to various kinetic models. The R2 values for zero-order kinetics ranged from 0.946 to 0.996, for first-order kinetics from 0.946 to 0.980, and for the matrix from 0.866 to 0.974. Korsmeyer Peppas kinetics had R2 values ranging from 0.935 to 0.935, while Hixon Crowell kinetics had R2 values ranging from 0.963 to 0.994. It was observed that SLN1, SLN3, and SLN8 showed Peppas, SLN2, SLN4, SLN6, and SLN7 showed zero-order kinetics, and SLN5 showed Hixon-Crowell. The experimental data were fitted to first-order, zero-order, Higuchi, Korsmeyer-Peppas, and Hixon-Crowell models in order to determine the mechanism of drug release. Zero order was found to have the highest association with the standardized formulation.
| Formulation code | Kinetic Models | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero-Order Plot | First-Order Plot | Matrix | Korsmeyer Peppas | Hix.Crow | Korsmayer peppas parameter | Best fit model | ||||||
| R2 | k | R2 | k | R2 | K | R2 | k | R2 | k | n | ||
| SLN 1 | 0.9973 | 1.5477 | 0.9340 | -0.0321 | 0.9292 | 9.9706 | 0.9974 | 0.9232 | 0.9716 | -0.0881 | 1.1386 | Peppas |
| SLN 2 | 0.9893 | 1.5269 | 0.9594 | -0.0299 | 0.9204 | 9.8267 | 0.9863 | 0.428 | 0.9795 | -0.0077 | 1.3427 | Zero order |
| SLN 3 | 0.9942 | 1.4260 | 0.9466 | -0.0264 | 0.9141 | 9.1394 | 0.9948 | 0.6075 | 0.9721 | -0.007 | 1.2252 | Peppas |
| SLN 4 | 0.9731 | 0.9077 | 0.9565 | -0.0124 | 0.8665 | 5.7313 | 0.9348 | 0.0027 | 0.9634 | -0.0037 | 2.5478 | Zero order |
| SLN 5 | 0.9759 | 1.3106 | 0.9695 | -0.0213 | 0.9300 | 8.5085 | 0.9351 | 0.4360 | 0.9788 | -0.006 | 1.3021 | Hix. Crow |
| SLN 6 | 0.9817 | 1.0168 | 0.9714 | -0.0142 | 0.9305 | 6.5890 | 0.9555 | 0.3758 | 0.9789 | -0.0042 | 1.2733 | Zero order |
| SLN 7 | 0.9898 | 1.2624 | 0.9711 | -0.0203 | 0.9146 | 8.1081 | 0.9816 | 0.8192 | 0.9810 | -0.0057 | 1.1102 | Zero order |
| SLN 8 | 0.9884 | 1.2826 | 0.9808 | -0.0204 | 0.9319 | 8.2979 | 0.9895 | 1.0713 | 0.9865 | -0.0058 | 1.0482 | Peppas |
| SLN 9 | 0.9962 | 1.2619 | 0.9659 | -0.0204 | 0.9183 | 8.0971 | 0.9351 | 0.7002 | 0.9814 | -0.0057 | 1.1545 | Peppas |
| SLN 10 | 0.9969 | 1.5335 | 0.9399 | -0.0312 | 0.9281 | 9.8775 | 0.9962 | 1.1742 | 0.9727 | -0.0079 | 1.0696 | Zero order |
| SLN 11 | 0.9839 | 1.5420 | 0.9562 | -0.0305 | 0.9302 | 9.9882 | 0.9588 | 2.1366 | 0.9748 | -0.0079 | 0.9087 | Zero order |
| SLN 12 | 0.9463 | 1.9483 | 0.9669 | -0.0603 | 0.9743 | 12.9129 | 0.9727 | 3.9166 | 0.9940 | -0.0125 | 0.8254 | Hix. Crow |
Table 4: Drug Release Kinetics with Best Fit Model of All Formulations of Felodipine.
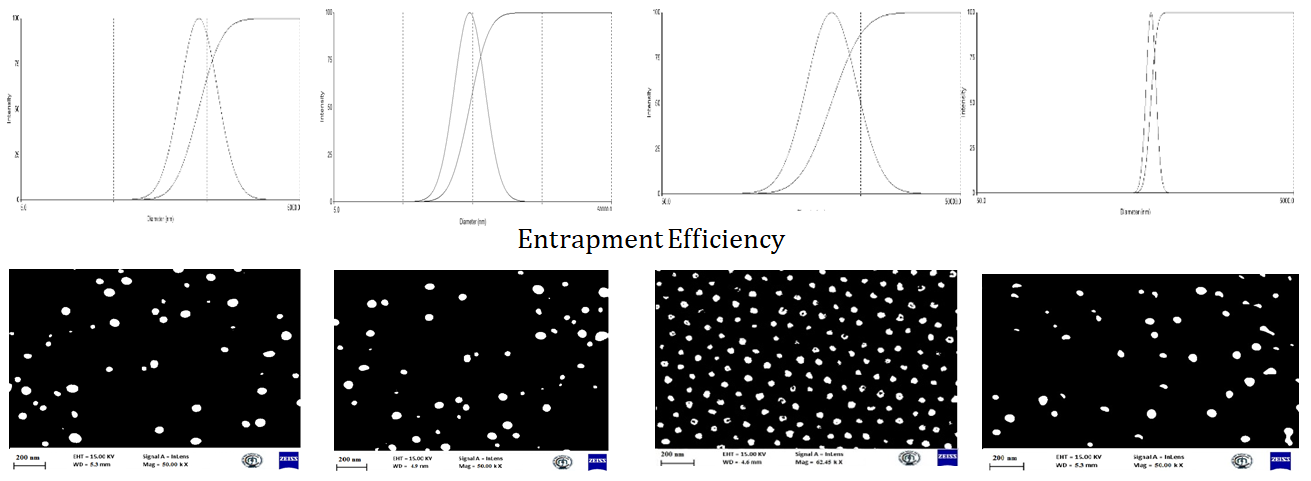
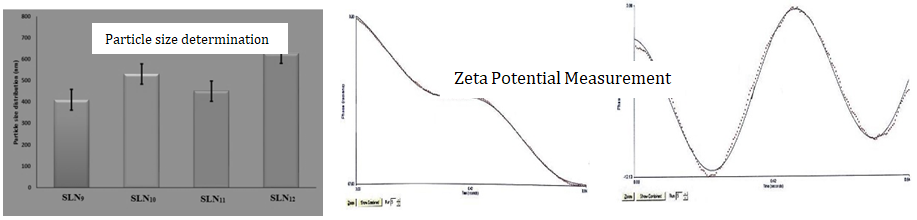
Evaluation of Optimization Formulation: Here, assay, entrapment efficiency, particle count by SEM, particle size distribution and polydispersity by DLS, zeta potential measurements, and in vitro drug release with a kinetic model are studied, as shown in Figures 4 & 5.
Predicted solution based on the optimization technique; the optimum target of drug release was fixed as 100% and optimization was carried out based on the results obtained from the initial 4 runs of the 22 full factorial designs. Based on the statistical data, Design Expert software generated the possible solutions, among those solutions one formulation was selected which is near to the maximum glycerol monosterate concentration which is having the desirability value of 1. (i.e., drug release =100%). Hence, the selected formulation based on 100% drug release is SLN13. Where in SLN13 the concentration of Glycerol Monosterate- 576.522mg and Poloxamer 188.
Evaluation of the optimized formulation (SLN13): Determination of particle size distribution and polydispersity by Dynamic Light Scattering, Zeta Potential Measurements (Figure 6) and Scanning Electron Microscopy, Assay, entrapment efficiency (Figure 7), in vitro drug release and kinetic studies are done (Figure 8).
| Time (min) | Abs(nm) | Conc. (µg/ml) | Amt. in sample (µg/ml) | TADR | DL (µg) | CDR (µg) | TDR | %DR |
|---|---|---|---|---|---|---|---|---|
| 10 | 0.129 | 2.6 | 13 | 2340 | - | - | 2.340 | 23 |
| 20 | 0.256 | 4.8 | 24 | 4320 | 13 | 13 | 4.333 | 43.7 |
| 30 | 0.379 | 7.4 | 36 | 6480 | 24 | 37 | 6.504 | 65.6 |
| 40 | 0.432 | 8.6 | 43 | 7740 | 36 | 73 | 7.776 | 78.5 |
| 50 | 0.483 | 9.6 | 48 | 8640 | 43 | 116 | 8.683 | 87.7 |
| 60 | 0.521 | 11 | 55 | 9900 | 48 | 164 | 9.948 | 100 |
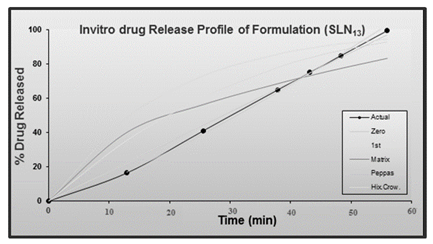
Table 5: In-Vitro drug release of the formulation (SLN13); Abs – Absorbance, Conc- concentration, TADR- Total amount of drug rele
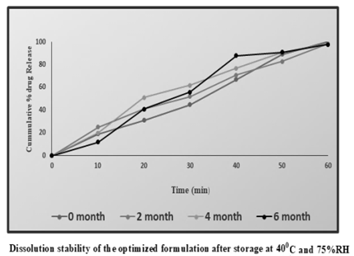
The chosen formulation came in aluminium foil packaging. For six months, they underwent stability investigations under accelerated testing (40-20°C/75- 5%RH) in the stability chamber. The sample’s physical and chemical properties were assessed at predetermined intervals of time. Physical characteristics: The capsules’ appearance, shape, hardness, brittleness, and flexibility were evaluated. Chemical parameters: As revealed in the drug content and dissolution experiments, formulations were estimated for the Figure 9.
| Time in Months | Physical Appearance | %Drug Content | % Drug Release in 60min |
|---|---|---|---|
| 0 | +++ | 99.88 | 100.4 |
| 2 | +++ | 98.90 | 98.34 |
| 4 | +++ | 98.74 | 98.56 |
| 6 | ++ | 98.68 | 97.86 |
Table 6: Shows the Study of Physical Parameters Assessed at Predetermined Intervals of Time.
Conclusion
The majority of drug delivery research focuses on nanotechnology-based methods to increase the bioavailability of medications with limited solubility. Due to the reduced particle size, the in vitro drug release testing for all formulations conducted at stomach pH 1.2 showed that the medication released immediately and within 60 minutes. In vitro release data were fitted to multiple kinetic models to understand the drug release process, and formulation results in better drug release. According to established ICH criteria, all nanoformulations underwent 6-month accelerated stability investigations, and it was found that neither the physical nor chemical appearance had significantly changed.
Conflicts of interest
No Potential conflict of interest
References
-
Steve IS, Bhaskara RJ, Xiaoling L (2003) New York: McGraw-Hills. Standard Handbook of Biomedical Engineering and Design pp: 22.1-22.2.
-
Mahammad RS, Madhuri K, Dinakar P (2012) Polymers in Controlled Drug Delivery Systems. International Journal of Pharmaceutical Sciences 2(4): 112-116.
-
Sunil K, Anil K, Vaibhav G, Kuldeep M, Pankaj R (2012) Oral Extended Release Drug Delivery System: A Promising Approach. Asian Journal of Pharmaceutical Technology 2(2): 38-43.
-
Chobanian AV, Bakris GL, Black HR, Cushman WC, Lee AG, et al. (2003) The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 289(19): 2560-2571.
-
Bhalla N, Deep A, Goswami M (2012) An overview on various approaches to oral controlled drug delivery system via gastroretentive drug delivery system. International Research Journal of Pharmacy 3(4): 128- 133.
-
Chugh I, Seth N, Rana AC, Gupta S (2012) Oral sustain release drug delivery system: An overview. International Research Journal of Pharmacy 3(5): 57-62.
-
Olbrich C, Gebner A, Kayser O, Muller RH (2002) Lipid– drug conjugate (LDC) nanoparticles as novel carrier system for the hydrophilic antitrypanosomal drug diminazenediaceturate. Journal of Drug Target 10(5): 387-396.
-
Muller RH, Runge SA (1998) Solid lipid nanoparticles (SLN) for controlled drug delivery, in: S Benita Submicron emulsions in drug targeting and delivery. Amsterdam: Harwood Academic Publishers 50(1): 161-177.
-
Jenning V, Gysler A, Schafer KM, Gohla S (2000) Vitamin A loaded solid lipid nanoparticles for topical use: occlusive properties and drug targeting to the upper skin. European Journal of Pharmaceutics and Biopharmaceutics 49(3): 211-218.
-
Jerzeweski RL, Chein YW (1992) Osmotic drug delivery in treatise on controlled drug delivery. International Journal of Pharmaceutical Sciences pp: 225-253.
-
Dunselman PH, Edgar B (1991) Felodipine clinical pharmacokinetics. Clinical pharmacokinetics. 21(6): 418-430.
-
Todd PA, Faulds D (1992) Felodipine: A review of the pharmacology and therapeutic use of the extended release formulation in cardiovascular disorders. Drugs 44(2): 251-277.
-
Ljung B, Nordlander M (1987) Pharmacodynamic properties of felodipine. Drugs 34(3): 7-15.
-
Singhal GB, Patel RP, Prajapati BG, Patel NA (2011) Solid lipid nanoparticles and nano lipid carriers: As novel solid lipid based drug carrier. International Research Journal of Pharmacy 2(2): 40-52.
-
Pardeike J, Hommoss A, Muller RH (2009) Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. International Journal of Pharmaceutics 366(1): 170-184.
-
Mehnert W, Mader K (2001) Solid lipid nanoparticles: Production, characterization and applications. Advanced Drug Delivery Reviews 47(2): 165-196.
-
Martins S, Sarmento B, Ferreira DC, Souto EB (2007) Lipid-based colloidal carriers for peptide and protein delivery-liposomes versus lipid nanoparticles. International Journal of Nanomedicine 2(4): 595-607.
-
Muller RH, Radtkeb M, Wissingb SA (2001) Solid lipid nanoparticles (SLN) and nanostructured lipid (NLC) in cosmetic and dermatological preparations. Advanced Drug Delivery Reviews 54(1): 131-155.
-
Madipatla PR, Manjunath K, Siddalingappa TB (2014) Development and evaluation of Bixin solid lipid nanoparticles for hepatoprotection. International Journal of Pharmaceutics 1(2): 485-492.
-
Jawahar N, Meyyanathan SN, Reddy G , Sood S (2012) Solid lipid Nanoparticles for oral delivery of poorly soluble drugs. Journal of Pharmaceutical Science & Research 4(7): 1848-1855.
-
Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, et al. (2000) Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. New England Journal of Medicine 342(3): 145-153.
-
Snyder BD, Rowland A, Polasek TM, Miners JO, Doogue M (2014) Evaluation of felodipine as a potential perpetrator of pharmacokinetic drug-drug interactions. European Journal of Clinical Pharmacology 70(9): 1115-1122.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State