Role of Ubiquilins in Neurodegenerative Diseases
Ubiquilin (Ubqln) proteins, including Ubqln1-4, are a family of ubiquitin-like (UBL) proteins that structurally contain anN-terminalubiquitin-like domain, a C-terminalubiquitin-associated (UBA) domain, and aninternal variable number of Stil repeat motifs. Unlike those small molecule ubiquitin-like proteins, Ubqln proteins cannot be covalently tagged to the proteins that are targeted to the proteasome for degradation. Instead, this family of proteins binds to thepolyubiquitinated proteins via the UBA domain and delivers them to the proteasome for degradation, despite their involving in autophagy-mediated protein degradation. Increasing studies have suggested that Ubqln proteins play important roles in mediating the degradation of damaged/misfolded proteins and thus theirinsufficiencies or dysfunctionsare frequently associated with different neurodegenerative disorders. This review summarizes the role of Ubqln proteins in several neurodegenerative disorders.
Introduction
Proteins are the most common macromolecules found in cells, including neural cells, and play a central role in various intracellular biological processes. However, proteins frequently become misfolded in various neurodegenerative disorders due to genomic mutations, transcriptional and translational failures, or various stress conditions such as oxidative stress. Since misfolded proteins generally lose their native confirmations and functions and preferentially tend to aggregate, they are potentially toxic to cells [1]. Cells have evolved two major mechanisms, the proteasome and autophagy pathways, dealing with misfolded proteins. This review focuses on the role of ubiquilin proteins in the proteasome pathway and several neurodegenerative disorders.
The Ubiquitin-Proteasome System (Ups)
The UPS is the major mechanism for degrading unneeded and damaged proteins in brain cells [2, 3, 4]. Degradation of a protein via UPS generally involves the following three steps: (1) covalently attaching a polyubiquitin chain to a protein substrate, (2) delivery of the polyubiquitinated protein to the 26S proteasome by ubiquilin (Ubqln) proteins, and (3) degradation of the polyubiquitinated protein by the 26S proteasome. The tagging of ubiquitin to target proteins occurs at the ε- amine of lysine residues of the proteins and requires a cascade of chemical reactions catalyzed sequentially by E1 (ubiquitin activating), E2 (ubiquitin conjugating) and E3 (ubiquitin ligating) enzymes [5] (Figure 1, lower panel). The repeat reactions by these three enzymes cause the target protein to be conjugated with a polyubiquitin chain that will function as a signal for targeting to the proteasome for degradation. Generally, the shortest ubiquitin chain recognized by the proteasome for degradation contains at least four ubiquitin moieties linked preferentially through lysine 48 (K48) [6]. The covalent ubiquitin bonds (isopeptide linkages) between ubiquitin and a target protein as well as those between ubiquitin proteins can be reversed by specific deubiquitinating enzymes (DUBs) (Figure 1). Once a polyubiquitin chain is added to a target protein, it will then be recognized bya ubiquitin receptor or adaptor, such as an Ubqln, which can deliver and couple polyubiquitinated proteins to the proteasome for degradation [7]. At the proteasome, the 19S lid of the proteasome contains deubiquitinating enzymes (DUBs) that function to remove the ubiquitin chain prior to the target protein entry into the cylinder-like 20S proteasomal core [8], where the target protein is degraded by the proteasome coreas it contains the protease active sites that perform the proteolysis reactions [9] (Figure 1, lower panel).
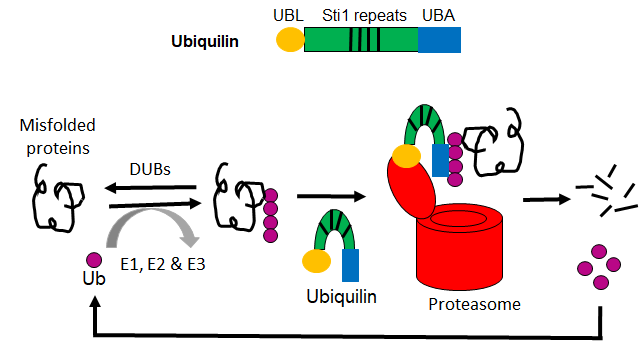
Top panel showing the schematic of Ublqn protein domain structure containing a UBL domain at the N- terminus, a UBA domain at the C-terminus, and a variable number of internal Stil repeats.
Lower panel showing the role of Ubqln proteins inUPS-mediated protein degradation. When a protein is misfolded, it is covalently conjugated with a polyubiquitin chain by the three enzymes, E1, ubiquitin activating enzyme, E2, ubiquitin conjugating enzyme, and E3, ubiquitin ligating enzyme. The ubiquitination step can be reversed by the deubiquitinating enzymes (DUBs). AUbqln protein binds to the polyubiquitinated protein via its UBA domain and delivers it to the proteasome where the Ubqln protein interacts with the 19S proteasome cap through its UBL domain. The DUBs located in the 19S cap of the proteasome recognize and break down the polyubiquitin chain into individual ubiquitin that will be recycled for ubiquitination reactions, while the target protein is cleaved into short polypeptides through the 20S proteasome core. Subsequently, the cytosolic peptidases further digest the polypeptides into amino acids that will be re-used in protein synthesis.
Ubiquilin(Ubqln) Family Proteins
Ubqlnsare a family of proteins characterized by having an N-terminal ubiquitin-like (UBL) domain, a C-terminal ubiquitin-associated (UBA) domain, and a variable number of internal Sti1 ( stress-inducible, heat shock chaperonin-binding motif) repeats [10, 11, 12] (Figure 1, top panel). Mammals have several Ubqln genes. Three of them, Ubqln1 (also known as PLIC-1), Ubqln2 (also known asPLIC-2), and Ubqln4 (also known as A1Up, UBIN, CIP75), are widely expressed in human and mouse, whereas Ubqln3 is only expressed in the testis in both human and mouse [10, 12, 13, 14]. The UBA domain of Ubqln proteins is responsible for interaction with polyubiquitin chain of ubiquitinated protein, while the UBL domain is considered to interact with the proteasome [7]. Accordingly, Ubqlns function as ubiquitin receptors or adaptors and deliver the polyubiquitinated proteins to proteasome for degradation [7]. Interestingly, some studies suggest that Ubqlns also regulate auto phagy- mediated protein degradation [15, 16]. However, the exact molecular mechanisms remain unclear.
Role of Ubqlns in Neurodegenerative Disorders
Several lines of studies have suggested that Ubqln1 is associated with Alzheimer’s disease (AD) and Huntington’s disease (HD). In late onset AD patient brains, Ubqln1 protein levels are significantly reduced and Ubqln1 immuno reactivity is associated with neurofibrillary tangle pathology [12, 17, 18]. Ubqln1 interacts and regulates both presenilin and amyloid precursor protein (APP) stability [19, 20, 21]. In the APP/PS1 AD mice, over expression (OE) of Ubqln1 reduces AD- caused learning and memory deficits and amyloid-β pathology [22]. The beneficial effect exerted by Ubqln1 OE is likely attributed to improved protein homeostasis in the brain cells, as Ubqln1 can selectively interact with misfolded proteins and enhance their degradation [23, 24]. Since AD is associated with oxidative stress and reduced UPS functionality [25, 26, 27], OE of Ubqln1 may be a therapeutic strategy for treating the disorder. Indeed, our studies strongly support this possibility [22]. In addition to AD, Ubqln1 is also implicated in HD. Ubqln1 is colocalized with mutant huntingtin inclusions in both cell and animal models [24, 28, 29]. OE of Ubqln1 promotes the turnover of mutant huntingtin and reduces mutant huntingtin cytotoxicity in cell models of HD [24, 30]. Consistently, OE of Ubqln1 in an HD mouse model increases the animal lifespan [31]. These data strongly suggest that an increase in Ubqln1 protein level is a therapeutic strategy for treating AD and HD.
In addition to the chronic neurodegenerative diseases, Ubqln1 also plays a neuroprotective role in acute brain injury such as ischemic stroke. OE of Ubqln1 protects mouse brains from acute oxidative stress and ischemic stroke-caused brain injury, while deletion of the gene exacerbates ischemic stroke-caused brain injury [32, 33]. In addition to improved UPS function, it is also possible that the beneficial effects observed from the chronic and acute neurological disorders by OE of Ubqln1 are caused by improved metabolism, as Ubqln1 transgenic mice also exhibited high levels of two energy-sensing proteins, sirtuin1 in the hypothalamus and AMP-activated protein kinase in the skeletal muscle [34]. Moreover, Ubqln1 itself appears to be an ATP-independent molecular chaperone protein at least for some plasma and mitochondrial membrane proteins [18, 23]. Thus, a sufficient amount of Ubqln1 should be necessary for maintaining these proteins’ normal structures and functions, especially in the acute or chronic oxidative stress conditions.
Compared to Ubqln1, Ubqln2 shows a more restricted expression pattern, indicating that it may play distinct roles from Ubqln1 in cells14. Indeed, one group of researchers has shown that Ubqln2 mainly mediates autophagy-independent protein degradation in the nucleus via the proteasome [35]. Mutations in Ubqln2 cause dominantly inherited, chromosome X-linked amyotrophic lateral sclerosis (ALS) with front temporal dementia [36, 37]. OE of the mutant gene in rodents mimic the human disease [38, 39, 40], possibly due to defective proteasome delivery of polyubiquitinated proteins by the mutant Ubqln2 [36, 41]. These results highlight the significance of the protein integrity in neuronal survival and normal functions.
Ubqln4 was originally identified as an ataxin-1 interacting nuclear protein [13]. However, increasing data suggest the protein playing significant roles in the cytoplasm. Lee, et al. showed that Ubqln4 functions as an adaptor protein to mediate the interaction between Ubqln1 and the autophagy machinery by recruiting Ubqln1 to LC3 [42]. Ubqln4 also interacts with BAG6, a UBA domain-containing protein [43], and recognizes misfolded proteins in the cytoplasm, targeting them to the proteasome for degradation [44]. More intriguingly, a variant inUbqln4 gene was found o be associated with ALS, whose expression impairs proteasomal function and compromises axon morphogenesis in mouse motor neurons and in zebra fish [45].
Conclusion
Ubqln family proteins play an important role in the UPS system by functioning as ubiquitin receptors and coupling the polyubiquitinated proteins to the proteasome for degradation. Emerging data also support that they involve in autophagy. Insufficiency or dysfunctions of Ubqln proteins are associated with several neurodegenerative disorders. A better understanding of the molecular mechanisms underlying Ublqn functions in normal and disease conditions would provide new insights into the pathogenesis of the several neurodegenerative disorders mentioned above, leading to identification of novel therapeutic strategies to these diseases.
References
-
Amm I, Sommer T, Wolf DH (2014) Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta 1843(1): 182-196.
-
Demartino GN, Gillette TG (2007) Proteasomes: machines for all reasons. Cell 129(4): 659-662.
-
Li X, Thompson D, Kumar B, DeMartino GN (2014) Molecular and cellular roles of PI31 (PSMF1) protein in regulation of proteasome function. J Biol Chem 289(25): 17392-17405.
-
Kim YC, Li X, Thompson D, DeMartino GN (2013) ATP binding by proteasomal ATPases regulates cellular assembly and substrate-induced functions of the 26 S proteasome. J Biol Chem 288(5): 3334-3345.
-
Schrader EK, Harstad KG, Matouschek A (2009) Targeting proteins for degradation. Nat Chem Biol 5(11): 815-822.
-
Ciechanover A, Stanhill A (2014) The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta 1843(1): 86-96.
-
Ko HS, Uehara T, Tsuruma K, Nomura Y (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett 566(1-3): 110-114.
-
Ehlinger A, Walters KJ (2013) Structural insights into proteasome activation by the 19S regulatory particle. Biochemistry 52(21): 3618-3628.
-
Tanaka K (2009) The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85(1): 12-36.
-
Wu AL, Wang J, Zheleznyak A, Brown EJ (1999) Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol Cell 4(4):619-625.
-
Marin I (2014) The ubiquilin gene family: evolutionary patterns and functional insights. BMC Evol Biol 14: 63.
-
Mah AL, Perry G, Smith MA, Monteiro MJ (2000) Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol 151(4): 847-862.
-
Davidson JD, Riley B, Burright EN, Duvick LA, Zoghbi HY, et al. (2000) Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein. Hum Mol Genet 9(15): 2305-2312.
-
Conklin D, Holderman S, Whitmore TE, Maurer M, Feldhaus AL (2000) Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis- specific gene that contains an ubiquitin-like domain. Gene 249(1-2): 91-98.
-
N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, et al. (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10(2): 173-179.
-
Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, et al. (2010) Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 19(16): 3219-3232.
-
Mizukami K, Abrahamson EE, Mi Z, Ishikawa M, Watanabe K, et al. (2014) Immunohistochemical analysis of ubiquilin-1 in the human hippocampus: association with neurofibrillary tangle pathology. Neuropathology 34(1): 11-18.
-
Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, et al. (2011) Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem 286(41): 35689-35698.
-
Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, et al. (2006) Ubiquilin 1 modulates amyloid precursor protein trafficking and Abeta secretion. J Biol Chem 281(43): 32240-32253.
-
El Ayadi A, Stieren ES, Barral JM, Boehning D (2012) Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63- linked polyubiquitination of lysine 688. Proc Natl Acad Sci USA 109(33): 13416-13421.
-
Massey LK, Mah AL, Monteiro MJ (2005) Ubiquilin regulates presenilin endoproteolysis and modulates gamma-secretase components, Pen-2 and nicastrin. Biochem J 391(3): 513-525.
-
Adegoke OO, Qiao F, Liu Y, Longley K, Feng S, et al. (2017) Over expression of Ubiquilin-1 Alleviates Alzheimer's Disease-Caused Cognitive and Motor Deficits and Reduces Amyloid-beta Accumulation in Mice. J Alzheimers Dis 59(2): 575-590.
-
Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ, et al. (2016) Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol Cell 63(1): 21-33.
-
Wang H, Monteiro MJ (2007) Ubiquilin interacts and enhances the degradation of expanded-polyglutamine proteins. Biochem Biophys Res Commun 360: 423- 427.
-
Liu Y, Hettinger CL, Zhang D, Rezvani K, Wang X, et al. (2014) The proteasome function reporter GFPu accumulates in young brains of the APPswe/PS1dE9 Alzheimer's disease mouse model. Cell Mol Neurobiol 34(3): 315-322.
-
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM (2008) Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29(11): 1607-1618.
-
Hope AD, de Silva R, Fischer DF, Hol EM, van Leeuwen FW, et al. (2003) Alzheimer's associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J Neurochem 86(2): 394-404.
-
Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, et al. (2004) Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett 571(1-3): 171-176.
-
Rutherford NJ, Lewis J, Clippinger AK, Thomas MA, Adamson J, et al. (2013) Unbiased screen reveals ubiquilin-1 and -2 highly associated with huntingtin inclusions. Brain Res 1524: 62-73.
-
Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, et al. (2006) Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet 15(6): 1025- 1041.
-
Safren N, El Ayadi A, Chang L, Terrillion CE, Gould TD, et al. (2014) Ubiquilin-1 over expression increases the lifespan and delays accumulation of Huntingtin aggregates in the R6/2 mouse model of Huntington's disease. PLoS One 9(1): e87513.
-
Liu Y, Lu L, Hettinger CL, Dong G, Zhang D, et al. (2014) Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci 34(8): 2813-2821.
-
Liu Y, Qiao F, Wang H (2016) Enhanced Proteostasis in Post-ischemic Stroke Mouse Brains by Ubiquilin-1 Promotes Functional Recovery. Cell Mol Neurobiol 37(7): 1325-1329.
-
Qiao F, Longley KR, Feng S, Schnack S, Gao H, et al. (2017) Reduced body weight gain in ubiquilin-1 transgenic mice is associated with increased expression of energy-sensing proteins. Physiol Rep 5(8): e13260.
-
Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, et al. (2016) UBQLN2 Mediates Autophagy- Independent Protein Aggregate Clearance by the Proteasome. Cell 166(4): 935-949.
-
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, et al. (2011) Mutations in UBQLN2 cause dominant X- linked juvenile and adult-onset ALS and ALS/dementia. Nature 477: 211-215.
-
Teyssou E, Chartier L, Amador MD, Lam R, Lautrette G, et al. (2017) Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging 58: 239.e11-239.e20.
-
Le NT, Chang L, Kovlyagina I, Georgiou P, Safren N, et al. (2016) Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD- linked UBQLN2 mutations. Proc Natl Acad Sci USA 113(47): E7580-E7589.
-
Huang B, Wu Q, Zhou H, Huang C, Xia XG (2016) Increased Ubqln2 expression causes neuron death in transgenic rats. J Neurochem 139(2): 285-293.
-
Ceballos-Diaz C, Rosario AM, Park HJ, Chakrabarty P, Sacino A, et al. (2015) Viral expression of ALS-linked ubiquilin-2 mutants causes inclusion pathology and behavioral deficits in mice. Mol Neurodegener 10: 25.
-
Chang L, Monteiro MJ (2015) Defective Proteasome Delivery of Polyubiquitinated Proteins by Ubiquilin-2 Proteins Containing ALS Mutations. PLoS One 10: e0130162.
-
Lee DY, Arnott D, Brown EJ (2013) Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep 14(4): 373-381.
-
Leznicki P, Roebuck QP, Wunderley L, Clancy A, Krysztofinska EM, et al. (2013) The association of BAG6 with SGTA and tail-anchored proteins. PLoS One 8: e59590.
-
Suzuki R, Kawahara H (2016) UBQLN4 recognizes mislocalized transmembrane domain proteins and targets these to proteasomal degradation. EMBO Rep 17(6): 842-857.
-
Edens BM, Yan J, Miller N, Deng HX, Siddique T, et al. (2017) A novel ALS-associated variant in UBQLN4 regulates motor axon morphogenesis. Elife 6: e25453.
- Origin, Evolution, and Functional Impact of Short Insertion- Deletion Variants in Human Genomes: A Review
- Harnessing Molecular Glues for Next-Generation Vaccine, Cancer and Cardiovascular Disease Drug Development: A Comprehensive Review
- Lateral Cervical Epidermal Inclusion Cyst in a Paediatric Patient: A Rare Case Report
- Malarial Plasmodium Falciparum with Hepatitis B and C Virus Infections among Blood Donors in Ife Central Local Government Area, Ile Ife, Osun State, Nigeria
- Withanolides and Withaferin A- What’s next in Ashwagandha Research
- Designing of Dual Pulse Photoacoustic Tomography for Imaging of Drug-Response and Tumor Growth