Origin, Evolution, and Functional Impact of Short Insertion- Deletion Variants in Human Genomes: A Review
Repetitive DNA is now recognised as crucial components of the genome landscape of several eukaryotes. The Short insertions and indels class of repetitive DNA account for approximately 12% of all variations in the human genome. Their unique genome organisation and architecture confer the DNA several unique characteristics which are avenues for DNA biophysical and gene regulation studies. Further, given their frequent occurrence in the genome they are used in several genome mapping efforts. The present review is an update on the indel variation in the human genome covering diverse section on their architecture, effects on genes and proteins and human diseases. Given their high density their roles in changes to meiotic biology and genome evolution assumes importance. With the availability of genome sequences of several mammals and model organisms along with parallel improvements in genotyping and bioinformatics tools indel are now applied to diverse fields of biology. The areas include wildlife biology, pathogen evolution, adaptation and disease prognosis. Indel-associated mutation occurs enhance the rates of point mutations through IDAM with implications for recombination and meiotic biology. The evolution of the olfactory-related and G-protein coupled receptor signalling genes are an important indicator to the role of indels in human genome. Indels enable virus adaptability and are source of evolutionary innovation. Several plant and animal trait mapping research frequently employ indels. In the area of medical genetics, they are used as prognostic markers. Several lineage tracing studies in mammals and ancient human studies have consistently used indels as marker due to their significant advantages. Parallel improvements in methods of genotyping and bioinformatics have facilitated indel genome research. Hence, this review is an update on this class of variation serving to diverse fields of biologists.
Introduction
Insertion–deletion mutations (indels) refer to insertion and/or deletion of nucleotides into genomic DNA and include events less than 1kb in length. This class of repetitive DNA are an abundant source of genetic variation accounting for ~12% of all variants in the human genome. This class of variation were less explored previously owing to the increased interest in other class of markers and lack of improved detection methods and bioinformatics tools. With the availability of the Human Genome sequences and sequences of model organisms the research community are now exploring various aspects of this class of variation. Their increased frequencies of association with human genes as reported in Human gene mutation database (HGMD) is also an indicator of their medical importance. The present review is targeted to cover several aspects of the indel marker starting from their genome organisation, structure-function relationship and impact on the genes and proteins. Their architecture and impact on transcription and translation of DNA and detection methods forms second and middle section of the review. Next, their association in several human diseases is covered briefly. The genetic and evolutionary impact of indels in covered in detailed to provide their roles in meiotic mechanisms, roles in adaptation and evolutionary effects. Their applications in diverse areas such as genome editing, as markers for plant and animal improvement, wildlife biology and biomedical applications form the last section of the review.
Repetitive DNA Classification and Structure
Current research suggests 50% of the human genome is made up of repetitive sequences, many of which are found within introns. Tandem repeats play a crucial role in telomeric protection and repeats make up 25–50% of the genomes of mammals in particular. Repetitive DNA sequences, satellite DNAs (satDNAs), and transposable elements (TEs) are crucial parts of the genome landscape. Nearly 50% of the human genome is composed of repeats, the transposable elements present in the protein-coding sections of 4% of human genes. Because they are located within introns, approximately 89.5% of repetitions have been assumed to be non-functional [1]. They play an important role in large genomes, such as the Drosophila and human genomes with a variety of functions such as genome function and evolution [2]. Traditionally, they are separated into two main categories: 1. transposable elements (TEs), which are found throughout the genome, and 2. satellite DNAs (satDNAs), which are made up of arrays of sequences repeated in tandem. satDNAs are predominant in the interstitial, sub telomeric, and pericentromeric chromosomal regions, where they form constitutive heterochromatin blocks. Class I (retrotransposons) and Class II (DNA transposons) are the two primary groups into which TEs are divided based on their transposition intermediates [2]. Since their functions were first unclear, SatDNAs and TEs are frequently referred to as the “dark matter” of the genome.
A particular DNA sequence that appears just once in the genome is called unique DNA. A DNA segment with a unique sequence that appears repeatedly throughout the genome is known as repetitive DNA. Ten to 105 copies of the sequence per genome are considered to be moderately repetitive DNA. Noncoding sequences contain the majority of moderately repetitive DNA. Many chromosomes have clusters of these repeats with higher concentration in the centromere and telomere regions. About 15% of human DNA is made up of satellite DNAs. Mega satellite DNAs are tandem repeat lengths of DNA that are 50–400 times longer and sequence around 1000 bp (l kbp) and are co-dominantly inherited. Repeated sequences between 14 and 500 bp in length are known as minisatellite DNAs ranging in length from 0.1 to 20 kbp. Numerous well-established research data demonstrate the importance of repetitive DNA in genome functions. To format the expression of distinct coding sequences and to arrange other tasks necessary for genome replication and precise transfer to progeny cells, generic repeating signals in the DNA are required. The cooperative molecular interactions that produce nucleoprotein complexes also depend on repetitive DNA sequence elements. Repeat elements make up a sizable portion of scaffolding/matrix attachment regions (S/MARs) and act as either initiators or boundaries for heterochromatin domains indicates they are components of the genome with key architectonic role in higher order physical structuring [3]. Since the repetitive DNA component is frequently larger than the coding sequence component this enables comparisons between sequenced genomes [4]. The discovery that repetitive DNA sequences that are highly polymorphic make up between 30 and 90 percent of the genome represents a significant advancement in genetic identification. As adaptable instruments, DNA-based molecular markers have established themselves in a number of disciplines, including genetic engineering, physiology, embryology, and taxonomy [5]. Microsatellites are multilocus probes that produce intricate banding patterns; they are often ubiquitous and non-species specific. They are co-dominant STMS (sequence tagged microsatellites) markers and dominant fingerprinting markers, forming an optimal marker system [6]. The ability to accurately characterize and classify repetitive DNA sequences and gain new insights into these crucial genomic components will be significantly advanced by the combination of rapidly developing sequencing technologies, ongoing development and improvement of bioinformatics tools and databases, manual curation, and adaptation based on the unique characteristics of each model organism of interest.
Organisation, Structure and Impact of Indels
Insertion-deletion mutations (indels) are defined as the insertion and/or deletion of nucleotides into genomic DNA and include occurrences shorter than 1kb in length [7]. Short indels account for approximately 12% of all variations in the human genome. A noteworthy finding from genome research is the positive association between indel frequencies at multiple genes, as documented in the Human Gene Mutation Database (HGMD). Consistent with this viewpoint, the probability of a particular gene/sequence to undergo microdeletion is proportional to its propensity for micro insertion. Microdeletions and microinsertions of 1 bp account for 48% and 66% of total variation, respectively. However, the incidence of residual lesions decreases as the DNA sequence length increases deleted or inserted [7]. Slippage mutagenesis, which involves the addition or removal of one copy of a mono-, di-, or trinucleotide tandem repeat, best explains many indels of greater than one base pair. Compared to mutations of various lengths, the frequency of in-frame 3-bp and 6-bp indels is shown to be substantially lower [8]. The heptanucleotide CCCCCTG, which shares similarity with the complement of the 8-bp human minisatellite conserved sequence/χ-like element (GCWGGWGG), is one of several sequence motifs that are discovered to be over-represented in the region of indels [9]. A glimpse of the mutational hotspot is provided by the overrepresentation of the previously identified indel hotspot GTAAGT and its complement ACTTAC in the vicinity of both indels [10]. DNA polymerases pause sites and topoisomerase cleavage sites are two additional motifs that are overrepresented in the area of microdeletions and micro insertions [11]. A consensus sequence that is determined to be remarkably similar to probable DNA polymerase arrest sites is identified in a number of micro- deletion hotspots (TGRRKM, or T-G-A/G-A/G-G/T-A/C). The deletion hotspot consensus sequence (TGRRKM) in reverse orientation is quite similar to the over-represented GTAAGT motif near indels [9]. Indels are “backbone mutations,” which can significantly alter the structure, stability, and function of DNA, whereas substitutions are “side chain mutations.” Changes in copy count (CCC) variations, which occur when one or more nucleotides are repeated or deleted, are the most common indels; they make up almost half of indels in natural genomes and are typically caused by polymerase slippage during DNA replication Topolska, et al. Also, indels are damaged DNA that have been imperfectly repaired. Damage, including double-stranded breaks, is caused by exogenous sources including radiation and toxins as well as by metabolic activities, transcription, and replication stressors that necessitate topoisomerase’s untangling of DNA [12]. This mechanism is inaccessible in nondividing cells, hence nonhomologous end joining (NHEJ), a different repair process, is employed. NHEJ can treat the majority of lesions and frequently results in 100% repair; if not, it causes minor (few base pairs) indels often flanked by small (1 to 2 bp) microhomologies [13]. Indel is thought to have started as a two-stage insertion/deletion process, where the wild-type sequence was changed into an intermediate and then into the final altered sequence in the second step. According to the currently accepted theory, an inverted repeat mediated the insertion process, which took place first and the inserted base was positioned after the bases to be deleted [14]. Two processes, 31Ins (D+I) Del and 35Ins (D+I)Del, have been attributed to the indel delGTAinsC, as seen in the example of the BRCA1 gene [GCCAAATGTAgtaTCAAAGGAGG] [15]. Reports of skewed data indicates to the addition or deletion of a comparatively modest number of bases. According to the dataset reported by Dai, et al. [16], the mutations leave the reading frame unaltered for 79 out of the 211 indels (37%). However, for most indels, a changed reading frame might increase the chance that the mutation an observation currently used in genetic clinics for diagnostics. According to Dittwald, et al. [17] inverted repeats have also been linked to the creation of micro-deletions and micro-insertions. The emergence or disappearance of conspicuous repeats as a result of the insertion or deletion of particular bases has a significant impact on Indel. A micro-deletion may result from the excision repair of a hairpin loop, which is made possible by an inverted repeat. According to inverted repetitions may also encourage accidental mispairing of the nascent strand, which leads to downstream sequence duplication. Table-1 Ratios of Deletions to Insertions in various genomes.
It has also been suggested that sequences with an axis of internal symmetry or that are symmetrical to another contiguous sequence on the same DNA strand upstream aid in the creation of secondary structure intermediates by facilitating deletions and insertions [18]. Frameshift (FS) and non-frameshift (NFS) are the two types of variations that can arise from insertions in coding regions. NFS indels, which are multiples of three base pairs, add or remove one or more amino acids without altering the remainder of the protein sequence. Whole genome sequencing of 250 families predicted a rate of 0.16 structural variations (>20 bp) and 2.94 indels (1–20 bp) each generation Tozaki When compared to the background secondary structure type distribution, NFS indels are highly enriched in secondary structure coil conformation (80%) and depleted in the helix (11%) and strand (9%). By correcting the open reading frame, FS indel may save a potentially harmful variant of the first FS indel. For instance, a 1 bp insertion saved a 2 bp insertion in the ARID3B gene(refs). The two variation sites have different protein sequences, though, if the two FS locations are far apart in the same protein. The CLTCL1 gene’s 1bp and 10bp insertions are an example (Lin 2017). Gene Ontology’s (GO) Biological Process and Molecular Function annotations, suggest significant detection of olfactory-related genes involved in sensory perception of smell, and G-protein coupled receptor signalling pathway enriched categories in genes related to FS indels [19]. Transcription-related biological processes are significantly more abundant in NFS instances. Coding indels are abundant in N- or C-terminal regions, according to studies on the impact of indels on protein structure. In NFS cases, higher N-terminal indels are recorded, while in FS cases, higher C-terminal indels [20]. Most protein sequences remain unchanged in both cases, suggesting that the structure and functionality of the impacted proteins are only slightly impacted. In summary the insertion and deletion of small fragments in the protein coil region may result in differences in binding affinity and gene expression, which in turn can drive evolution and contribute to the diversity of phenotypes.
Repetitive DNA Mediated Replication and Recombination
Eukaryotic genomes contain many repetitive DNA sequences such as indels that exhibit size instability. The repeats form secondary structures, such as hairpin loops, slipped DNA, triplex DNA or G-quadruplexes [21]. The indel sequences mediate replication and recombination through a variety of molecular mechanisms often involving secondary DNA structures, mispairing and the recruitment of DNA repair and recombination repair [22]. When repeat sequences are longer as altered by indels, the DNA structures can form a significant impediment to DNA replication and repair, leading to DNA nicks, gaps, and breaks. Repair or replication fork restart attempts within the repeat DNA can lead to addition or removal of repeat elements Erica, et al. Alteration in replication fork progression is a defining feature of replication stress and the consequent failure to maintain fork integrity and complete genome duplication within a single round of S-phase compromises genetic integrity” [23]. One important DNA repair mechanism to maintain genomic integrity is recombination. Damages that result in DSBs can be repaired by various types of end-joining (EJ), by annealing of processed ends, or by recombination-based mechanisms using either a sister chromatid or homolog as the template. Due to the challenges of aligning DNA across a repetitive sequence, gain or loss of repeat units can occur during both homologous recombination (HR) and EJ [24]. In addition, recombination is a primary mechanism used in restarting stalled or collapsed replication forks and in repairing gaps left behind the replication fork DNA repair pathways protect the genome and maintain genome integrity, however indels in DNA can lead to inappropriate repair, repeat instability, or genome rearrangements [25]. Chromosomal DNA replication is essential to maintain genomic stability however indel repetitive sequences impair replication, which leads to replication slippage, secondary structure formation, replication fork stalling and genomic instability [26]. One of the earliest proposed mechanisms for contractions or expansions of repeats was replication slippage a process involving the template and nascent strands reannealing out due to the repetitive nature of the template [27]. DNA polymerase slips during replication, particularly at indels, resulting in insertion and deletion of DNA sequence. According to Khristich and Mirkin, some of these expandable repeats (mono-, di-, and trinucleotide) can form stable non-B- form DNA structures (secondary structures) such as hairpin loops, cruciforms, G-quadruplexes or triplex structures which can stall replication fork, causing fork collapse and template switching results in copy number variations. In addition to fragility, replication problems at structure forming repeats can also lead to repeat expansions and contractions [28]. HR and EJ can be mutagenic when they occur within repetitive DNA, resulting in a loss (contraction) or gain (expansion) of repeat units [29]. Figure 1 illustrates indel induced replication slippage. Non-allelic homologous recombination (NAHR) occurs through the same fundamental mechanism in meiosis as (HR) except that the pairing of the homologous chromosomes is non-allelic and occurs between misaligned repetitive sequences such as segmental duplications (SDs) that are present in the genome. When this happens, sequences that lie between the repeats that undergo NAHR will be either deleted or duplicated, thus changing the copy number. Of note here is that NAHR-mediated deletions and duplications will arise when the SDs are in the same orientation [30]. NA double-strand break (DSB) repair through direct ligation by non-homologous end joining (NHEJ), whereas long stretches of sequence homology at or near the breakpoint are repaired by HR [31, 32]. HR repairs DSBs using template sequences and relies on the presence of DNA segments sharing extremely high similarity or identity [33].
Detection of Indel-Laboratory and Computational Methods
The accurate detection of short indels is fundamental to the understanding of the origin, evolutionary dynamics and functional consequences in the human genomes. Detection of indels smaller than 50 base pairs, have become a central objective in human genomics due to their substantial contribution to genetic diversity and functional variation. After single nucleotide variants (SNVs), short indel represent the second most prevalent class of genetic variation in the human genome. Indels alter local sequence architecture and shift alignment coordinates, making their identification more computationally complex and error-prone [34, 35]. Over the past two decades, advances in molecular biology, high-throughput sequencing and computational genomics have substantially improved detection accuracy, scalability and interpretability.
Initially, indel detection relied on Sanger sequencing (gold standard for validating short indels due to its high base accuracy and direct readout of modified sequences) and capillary electrophoresis, which provided high accuracy but lacked scalability. However, RNA sequencing (RNA-Seq) has added an important functional dimension to indel detection by capturing expressed variants. Few shortcomings of detection methods include short reads frequently misalign around indel breakpoints, generating false positives or masking true variants due to local alignment ambiguity [34]. Present tools designed for variant detection in transcriptomic data, such as RNAIndel used in tumor biology, which perform realignment and incorporate machine-learning models to differentiate real coding indels from artifacts. Transcriptomic evidence is particularly useful for identifying biologically active indels that influence splicing, cause frameshifts, or alter allele-specific expression, complementing the DNA-based discovery. The emergence of next-generation sequencing (NGS) technologies has transformed variant discovery by enabling massively parallel sequencing at population scale [36]. However, short read lengths characteristic of early NGS platforms introduced substantial challenges in accurately identifying indels, particularly within repetitive or low-complexity genomic regions [37]. To address these challenges, newer computational strategies have evolved considerably since the last decade. Early alignment- based variant callers such as SAMtools and the Genome Analysis Toolkit (GATK) introduced probabilistic models to distinguish true variants from sequencing errors [38]. Dindel (Detection of INDELs by realignment) further improved small indel detection by explicitly modelling candidate indels and realigning reads locally [34]. These developments have marked a shift from simple pileup-based detection to local realignment and likelihood-based frameworks, substantially improving sensitivity and specificity. Split-read and paired- end mapping approaches have further enhanced breakpoint resolution by leveraging discordant alignments [39]. In parallel, local de novo assembly strategies reconstructed variant sequences within targeted regions, have reduced alignment artifacts and improved detection of complex or clustered indels Assembly-based callers such as Platypus and Scalpel have demonstrated improved performance in regions with high sequence complexity by reconstructing haplotypes [40]. Recent advances in long-read sequencing platforms, including Pacific Biosciences and Oxford Nanopore technologies, have improved detection of medium and complex indels by spanning repetitive regions and reducing alignment ambiguity [41]. Although historically associated with higher raw error rates, improvements in consensus accuracy (e.g., HiFi reads) have significantly enhanced small variant detection performance. Finally, Hybrid approaches combining short- and long-read data have further increased reliability and completeness in variant catalogs.
Population-scale sequencing projects have expanded the catalog of short INDEL variation and clarified their evolutionary dynamics. The 1000 Genomes Project demonstrated that short indels exhibit heterogeneous mutation rates and are subject to stronger purifying selection in coding regions compared with many SNVs Functional analyses have shown that frameshift indels can have profound phenotypic consequences, particularly in regulatory and protein-coding sequences [42, 43].
Collectively, accurate detection of short indels requires an integrated framework combining advanced sequencing technologies, probabilistic alignment models, local assembly strategies, and rigorous benchmarking. Despite substantial progress over the past two decades, challenges persist in repetitive genomic regions, in insertion modelling, and in cross-platform concordance. Ongoing methodological refinement and multi-technology integration remain essential for improving sensitivity, precision, and functional interpretation of short indel variation.
Indels Associated with Human Disorders
Insertions and deletions, also known as indels, are changes in the DNA sequence that involve adding or removing one or more nucleotides. These types of changes are very common in the human genome, ranking second in frequency after single nucleotide polymorphisms (SNPs). They account for 15 to 21 percent of all genetic variations in humans. When indels occur in regions of DNA that code for proteins, they can lead to two distinct types of genetic changes: frameshift mutations and non-frameshift mutations Lin, et al. Several of these are associated with diverse Human disorders. We provide few examples in the following paragraph covering various pathologies.
In neuropsychiatric conditions there have been no general reports of a significant burden of rare structural genomic variants such as indels in bipolar disorder (BD), however, few individual cases involving microduplication and microdeletion have been documented. BD is a long-term, multifactorial mental health condition that impacts mood, thinking, and daily functioning [44]. Singleton deletions (rare copy number variants (CNVs) account for 16.2% of the BD cases studied [45]. Autism spectrum disorder (ASD) is a type of neurodevelopmental condition the genetic correlates of which are yet completely understood. A study by Wongpaiboonwattana, et al. [46] sought to explore possible links between ASD and a 19-base pair insertion or deletion variation in the dopamine beta-hydroxylase gene (DBH), which is important for the processing of neurotransmitters. 19-bp insertion allele was significantly higher in the patient group compared to the controls (p = 0.046). The clinical symptoms caused by GFAP (glial fibrillary acidic protein) mutations such as the Y349Q350insHL indel in the C-terminal and central rod domains show a wider range in the age when symptoms first appear and the main neurological signs. These include spastic paraparesis, cerebellar ataxia, and less commonly, symptoms resembling progressive supranuclear palsy (PSP) [47]. A study conducted by Lin, et al. [48], to examines the role of the ACE I/D polymorphism in individuals with Alzheimer’s disease, focusing specifically on its connection to hypertension brain volume. The study suggests no significant differences in ACE genotypes and the presence of the apolipoprotein epsilon 4 (APOEε4) allele, or brain volume. A novel indel mutation in ryanodine receptor 1 (RYR1) is shown to be associated with mild calf-predominant myopathy Jokela, et al. In Parkinson’s disease a 18 bp promoter variant of the DJ-1 gene alters REST transcription factor binding and regulates its expression.
Osteogenesis imperfecta (OI) type I, which is caused by the null allele of the COL1A1 gene, is the most common form seen in clinical cases. In heterozygous Mov-13 mice, partial knock out of exons (exon2 – exon 5, 365 nt of mRNA) using CRISPR/Cas 9 system, resulted in large decrease in type I collagen synthesis due to frame shift mutation and premature chain termination, mimicking pathogenic mechanism in affected individuals. The strain showed greatly reduced mineral structures in the bones, along with bone loss, decreased mechanical strength, indicating a continuous weakening of the skeletal system [49]. Diabetic kidney disease (DKD) is a frequent microvascular complication that occurs in diabetes, and its development involves multiple factors, including genetic influences. Growth arrest-specific 5 (GAS5) is a long noncoding RNA (lncRNA) gene with roles in renal functions. An insertion/deletion polymorphism (rs145204276) is shown to be associated with phenotype fibrosis [50].
Indels are associated with kidney diseases. In autosomal- recessive polycystic kidney disease a indel mutation causes a frameshift within Pkhd1 gene exon 48, resulting in a premature termination codon (UGA) Yang, et al. Filaggrin, a protein produced by the FLG gene found in the epidermal differentiation and is essential for skin function. It enables development of the stratum corneum the outermost layer of the epidermis. Yuda [51] identifies single nucleotide variants, insertion-deletion variants, and CNVs, including several loss- of-function mutations.
In cardiac diseases a study carried out by Temel [52], to examine the connection between ACE indel polymorphism and coronary artery disease (CAD) in Turkish Cypriots, indicating an association between ACE and CAD. 12 indel across six mitochondrial calcium uniporter (MCU) complex genes are reported to be associated with genetic predisposition and Mitochondrial dysfunction in Sudden Cardiac Death syndrome [53]. Further, in retinal diseases genetic mutations in the retinol dehydrogenase 5 (RDH5) gene are linked to inherited retinal degeneration conditions that follow an autosomal recessive pattern, especially fundus albipunctatus (FA). Both RDH5/WT and RDH5/L310delinsEV were found to interact with the autocrine motility factor receptor (AMFR), which functions as an E3 ligase located on the endoplasmic reticulum. Overexpression of AMFR or its knockdown by siRNA leads to an increase or decrease in the degradation of RDH5/L310delinsEV, respectively suggesting their roles in the transcription [54]. A novel indel CYP1B1 variant in a large multigenerational Pakistani family is reported in primary congenital glaucoma. Several indels are associated with several types of cancer and have aided in the clinical diagnosis. Hepatocellular carcinoma (HCC) is one of the most common types of cancer globally. The growth arrest specific 5 (GAS5) is known to play a role in different types of cancer. Tao [55] evaluated the association of a 5-base pair indel polymorphism (rs145204276) in the promoter region of GAS5 gene with HCC susceptibility in Chinese populations. Mutational signatures are increasingly used to understand the mechanisms causing cancer. However, their important applications are in predicting prognosis and stratifying patients. To this end few examples suggest application of indels such as the 18-bp indel GABPα which inhibits tumor progression and angiogenesis within VEGF promoter in breast cancer [56]. Combined single-base substitution (SBS) and indel (ID) spectra analysis, method has enabled accurate identification of various DNA repair deficiency signatures and patient survival prediction in high-grade serous ovarian cancers (HGSOC) [57]. Table 2 lists few indels associated with human diseases (Tables 1-4 & Figure 1).
Genetic and Evolutionary Implications of Indels
Indels cause local genetic variations that prevent correct crossover formation during meiosis, resulting in reduced genetic exchange in the immediate neighborhood. They also disturb homologous alignment, which frequently results in decreased crossover activity within the altered region. Several processes via which indels contribute to genomic alterations include a. Local suppression mechanism in which the indels cause a considerable decrease in crossing frequencies in the immediate and surrounding region. Kvikstad [58] conduct wavelet-based analysis of themes linked with DNA pol activity, topoisomerase cleavage, double stranded breaks (DSBs), and their repair, stressing the distinction between insertions and deletions. The model shows that indel mutagenesis includes both replication and recombination.
| Organism | Insertion | Deletion |
|---|---|---|
| Chimpanzee | 28.63% | 26.54% |
| Rhesus | 32.52% | 32.50% |
| Mouse | 35.31% | 40.87% |
| Tree shrew | 40.53% | 45.93% |
| Guinea pig | 40.50% | 42.21% |
| Rabbit | 71.00% | 46.74% |
| Cat | 43.33% | 43.19% |
| Cow | 38.38% | 42.67% |
| Elephant | 41.78% | 44.28% |
Table 1: Ratios of Deletions to Insertions in various genomes.
| Gene | Type of Indel | Molecular Consequence | Associated Disease(s) | Key Mechanism | Reference |
|---|---|---|---|---|---|
| CFTR | 3-bp deletion (ΔF508) | Loss of Phe508; protein misfolding | Cystic fibrosis | Defective chloride transport | Riordan |
| BRCA1 | Frameshift deletions | Premature stop codon; truncated protein | Breast & ovarian cancer | Defective homologous recombination repair | Miki |
| BRCA2 | Frameshift INDELs | Loss of functional BRCA2 protein | Breast, ovarian cancer | Impaired DNA repair | Wooster |
| APC | Frameshift deletions | Truncated APC protein | Familial adenomatous polyposis, colorectal cancer | Dysregulated Wnt signaling | Groden |
| EGFR | deletion-insertion | EGFR exon 19 deletion- insertion | Lung Cancer | tumor progression and metastasis. | Zhang R |
| HBA2 | deletion-insertion | point mutations (single nucleotide substitutions) | β Thalassemia | fetal and adult hemoglobin (HbA) that transports oxygen. | Das R, |
| ACE | Insertion/Deletion | Left Ventricular Hypertrophy in Patients with Hypertension | hypertension | Li | |
| PDE6B | c.1923_1969delins TCTGGG deletion | frameshift indel | retinal degeneration, | recessive rod-cone degeneration and autosomal dominant congenital stationary night blindness | Sangermano |
| ALDH1A1 | insertion/deletion | Alu transposon | Parkinson’s disease. | suppressive activity on gene transcription. | |
| ACE | insertion/deletion | - | Henoch-Schonlein purpura nephritis | an inflammation of the small blood vessels in the kidneys (glomerulonephritis) | Yan |
| KRT4 | insertion/deletion | amino acid mutation involving glycine. | White Sponge Nevus (WSN) | oral mucosa affected | Liu |
| Jiang 2008 | Jiang 2009 | Jiang 2010 | Jiang 2011 | Jiang 2012 | Jiang |
| Human Populations | No. of Indels | Method | Indel Size Range (Bp) | Rate(%) | |
| Mullikin, et al. 2000 | NR, not reported; | ABI trace mapping | NR | N/A, not applicable | |
| Bhangale et al. 2005 | 2393 | PCR/sequencing | 1–543 | N/A, not applicable | |
| Mills et al. 2006 | 415 436 | ABI trace mapping | 1–9989 | 97 | |
| Kim R. N 2012 | 4 genes reported | Sanger sequencing | 15-Mar | N/A, not applicable | |
| 1000 Genomes Project Consortium. 2015 | 1,000 large deletions, | 1,000 large deletions, | Not reported | N/A, not applicable | |
| Anders Bergström 2020 | 8.8 million small insertions or deletions | Whole genome sequencing (WGS) | Not reported | N/A, not applicable | |
| Alba Sanchis-Juan et al., 2023 | 88% of the total variation | Next gen sequencing (NGS) | Not reported | N/A, not applicable | |
| Shunichi Kosugi & Chikashi Terao 2025 | Not reported | Next gen sequencing (NGS) | 6- 50 bp, | N/A, not applicable | |
| Personal human genomes | |||||
| Wang et al. 2008/Han Chinese | 135 262 | Illumina/SOAP | 1–3 | 90–100 | |
| Schuster et al. 2010/ African | Not reported | N/A, not applicable | N/A, not applicable | N/A, not applicable | |
| Amalio Telenti 2016 | 25 genomes 0f African, European, and Asian, and admixed individuals | Next gen sequencing (NGS) | Not reported | N/A, not applicable | |
| Ganesh et al., 2026 | Indian descent from the state of Karnataka (KGP), | NGS | Not reported | N/A, not applicable | |
| Zimin,2022 | Puerto Rican | NGS | Not reported | N/A, not applicable | |
| Shumate | Ashkenazi | NGS | Not reported | N/A, not applicable |
Table 2: List of few genes with indels and the associated phenotype.
| HGMD Ref. no./Gene | Num Del | Num Ins | Type of Repeats | Proposed Path |
|---|---|---|---|---|
| CX992097/ ABCA4 | 6 | 4 | 55 | Del Ins |
| CX972728/APC | 1 | 3 | 34 | Del Ins |
| CX983261/ ATM | 3 | 4 | 53 | Ins(I+D)Del |
| CX900304/BCHE | 1 | 2 | 54 | Ins(D+I)Del |
| CX972734/CDKN1C | 2 | 1 | 44 | Ins(I+D)Del/ Ins(D+I)Del |
| CX921033/ DMD | 2 | 2 | 11,51/11 | Ins(I+D)Del/ Ins(D+I)Del |
| CX972791/ GLA | 1 | 3 | 9 11 | Del Ins |
| CX962381 / LPL | 4 | 2 | 33 | Ins(D+I)Del |
| CX941939 / PCCB | 1 | 4 | 12 41 | Del Ins |
| CX962712/ VHL | 2 | 2 | 53 | Ins(I+D)Del |
Table 3: Representative list of genes with possible mechanism and path of Insertion/Deletion events.
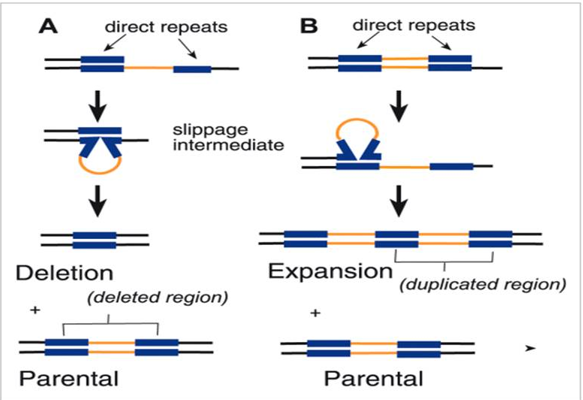
b. Recombination rejection-in this mechanism, indels within recombination intermediates cause mismatches, resulting in heteroduplex rejection and non-crossover (NCO) repair rather than crossover. Ziolkowski [59] investigate the effect of meiosis on genetic variation, arguing that sequence polymorphisms can feed back into recombination pathways. The work shows that heterozygous sequence polymorphisms can alter meiotic recombination pathways in both cis and trans c. Impact on Hotspot- insertions and deletions, are frequently associated with local cross-over (CO) suppression. Indels limit crossover frequency in strong recombination hotspots, resulting in a significant decline in local (CO) rates. Szymanska-Lejman, et al. [183] study on DNA polymorphisms and natural variation on crossover hotspot activity in Arabidopsis hybrids examines the effect of variation adjacent to meiotic hotspots on recombination. The study found that a modification of <7 kb around hotspots did not significantly impact their activity. Indels can also diminish recombination locally, and they are known to produce topological constraints for homologous pairing, resulting in a lower frequency of recombination enabling the genetic isolation of the two haplotypes. Mutations accumulate in each haplotype throughout time, resulting in a highly divergent indel-linked dimorphism [60, 61]. In addition, the enhanced mutation rate around indel may hasten the accumulation of dimorphic substitutions. Thus, nucleotide polymorphisms caused by point mutations could be preserved in the deletion junction regions between haplotypes. Indel-associated mutation occurs when indels boost the rate of point mutations in the surrounding DNA a phenomenon known as (IDAM) or indel- associated polymorphism [62]. Heterozygous indels produce mispairing during meiosis, which activates error-prone DNA repair machinery, resulting in nucleotide changes at the mutation site. Guo’s findings in arabidopsis found a dimorphic pattern with highly divergent areas surrounding 18 analyzed indels, and indels were related with four known dimorphic loci. Furthermore, heterozygous indels have been shown to be more mutagenic than homozygous indels because they lead chromosomes to produce mismatched heteroduplex structures during meiotic recombination or DNA repair. When a chromosome with heterozygote tries to couple with its homologous counterpart (which is wild- type or has a different indel) during meiosis, non-alignment results in a “loop” or bubble of DNA. Coelho [63] found that heterozygous mutations can promote genomic instability in a yeast model of cancer evolution. The genetic code is read in triplets (codons), therefore polymorphisms such as indels cause insertion or deletion of nucleotides that are not divisible by three shifts in the mRNA sequence’s “reading frame” [64]. Examples include the missense polymorphisms involved in mandibular prognathism [65] and truncating mutations within the COL6A1 gene in Ullrich congenital muscular dystrophy [66]. Indels are often destabilizing substrates in gene conversion since they introduce sequence mismatches that might initiate repair, prompt heteroduplex rejection, or cause frameshift mutations [67]. Frameshift variants, especially minor indels, impair protein coding and play an important role in human disease development. If these events do not occur in multiples of three during translation, the entire amino acid sequence may be disrupted. This disruption could cause loss of function (LOF), nonsense variants, or structurally faulty proteins, all of which have consequences in disorders such as like Duchenne muscular dystrophy (DMD), cystic fibrosis (CF), and hereditary breast cancer [68]. Similarly, Lalonde [69] report a segregating nonsense variant (rs2273865) located in a “multiple of three nucleotides” in the exon of LGALS8 gene that increases exon skipping in human erythroblast samples. Similarly, Chen [70] report structural and functional analysis of indels in the somatic coding and UTR regions in genes implicated in breast and lung cancer genomes.
It is not clearly known to what extent natural selection could have affected signatures of hotspots and motifs in the vicinity of indels identified in genic sequences. However, the scale at which a hotspot or a motif can be detected and implicated in a particular molecular process constitutes an active area of research. Additionally, combinations of specific binding sites acting jointly to promote transcription the simultaneous presence of multiple motifs could provide important clues to indel mutagenesis and requiring investigation [71]. Heterogeneity in base composition and substitution rate along the genome may have confounding effects on the results. Finally, using a multiscale methodology to analyze sequence contexts around a genome-wide set of indels found outside of genes is expected to contribute in determining the biological mechanisms driving these mutations [72]. In terms of selective pressures acting on indels, deletions consistently segregate at lower rates than insertions, both within genes and across the genome, implying that deletions are subjected to higher purifying selection. A mechanistic explanation is that deletions have two breakpoints while insertions only have one, making them more likely to strike a critical motif.
The difference in mean allele frequencies between the two categories of variation has also been explained by selection acting on concordantly. A number of studies have inferred higher fixing rates for insertions by comparing the ratio of deletion to insertion events between polymorphism and divergence data. This fixation bias is consistent with a number of hypotheses, including selection on insertions to preserve intron lengths and insertion-biased gene conversion. However, studies show the presence of mutation hotspots in repetitive regions and cryptic hotspots in non- repetitive regions, which could explain fixation biases by increasing rates of ancestral state misidentification [73]. Research data reveals that differences in the rate of ancestral misidentification between polymorphism data and divergence data and insertions found that they were especially vulnerable to misleading fixation signals [74]. As an example of this mechanism show that genome-wide indel appear to have negative consequences [75], with most coding indels severely deleterious and a sizable minority of noncoding indels displaying purifying selection signs in the Great Tit (Parus major) genome. The study also found that noncoding indel diversity is limited by connection to certain locations near exons and in low recombination regions.
Purifying selection is common across the genomes of several species, hence variants affected by purifying selection will, on average, tend to be present at lower frequency than expected under neutrality (i.e., the site frequency spectrum (SFS) will depart from the neutral expectation [76]. Compared to SNPs, short indel, especially those in functional genomic regions, are considered more likely to cause fitness reductions and to be under purifying selection [77]. In line with these expectations, the difference in nucleotide diversity between non-coding and coding SNPs has been found to be smaller than the difference between non-coding and coding indels [75, 78]. In model organisms, short indel were also found to be segregating at lower frequencies with a clear excess of low-frequency alleles for deletions in coding regions, as indicated by the most negative values of Tajima’s D [79]. These two patterns are strong pieces of evidence for purifying selection acting on short indels.
Microsatellites, for example, exhibit a “birth and death” process in which short, repetitive sequences expand through insertions and are controlled by deletion bias [80]. Their influence varies from rapid functional modifications to long- term genomic size evolution. This process has resulted in significant amounts of lineage-specific DNA in mammalian genomes, including the genome of Homo sapiens [81]. The birth-and-death model is also used to describe the evolution of multigene families, in which gene duplication (birth) is followed by functional divergence or deletion (death), frequently leading in the formation of pseudogenes like the HOX gene [82]. Indels are generally deleterious, but they are also potent engines of evolution and disease progression. In cancer, indels within tumor suppressor genes frequently cause frameshifts, resulting in premature stop codons and a lack of function that allows cells to grow uncontrolled. An example is a frameshift mutation in the APC gene that causes colorectal cancer [83]. Indels in viruses are important for adaptability because they serve as a primary source of evolutionary innovation, allowing viruses to adapt to new hosts, evade immunological responses, and acquire treatment resistance. Indels provide a “high-risk, high-reward” method, for generating more significant changes to protein structure and function. They also allow for adjustments to surface proteins or changes in enzyme performance, thereby driving the evolution of greater virulence [84, 85].
Indel is a unique evolutionary event that is unlikely to recur in the same location in several, unrelated lineages, making it an excellent sign of shared ancestry and evolutionary branching points. An illustration is the study by Paśko [86] who found a molecular consensus tree in 29 families of neognathous birds, supporting the correlation between indel fixation rates and lineage-specific evolutionary rates. According to the study, Galloanseres fixation rates was 1.5 times greater than Neoaves, and 2.4
times higher in the Rallidae than the average for Neoaves (8.2 times higher than the allied Gruidae). Because indel mutations are rarer than point mutations, sharing a specific indel typically indicates a common ancestor, making them useful markers for tracing species divergence, population history, and connections in humans and other species. In mammals, indels are around 14 times less prevalent than nucleotide changes [87]. Indel-based techniques have allowed to access evolutionary distance of the Mouse- Human Divergence indicating a major, deeply entrenched divergence, with high average indel rates in genome comparisons. Thus, indel-based evolutionary distance analysis contributes to the refinement of the Tree of Life, particularly when working with closely related genomes, by offering a supplementary, and frequently more stable, approach. The substitution-to-indel rate ratio is closer to 8, indicating stronger indel activity than previously anticipated [88]. The evolutionary distance, or the number of mutations fixed since the last common ancestor, is approximately 0.8 substitutions per site. Indels have enabled track species divergence and determine evolutionary links between humans and primates. Furthermore, they are reliable phylogenetic markers because they are less susceptible to convergence (homoplasy) than single nucleotide alterations, making them useful for reconstructing evolutionary histories. Chen [89] conducted a study on indel variation in or near genes and evaluated if this variation is significantly connected with previously reported differences in gene expression between humans and chimps. The data show that big indels (80 to 12,000 bp), notably those associated with retrotransposons, have contributed significantly to changes in gene regulation. In another study, Elizabeth [90] discovered 21,269 non-polymorphic indel insertions in the human genome. 372 indels of these were exon-specific human-specific insertions that were not observed in five comparative primate species: chimp, gorilla, orangutan, gibbon, and macaque. The remaining 20,897 are expected to provide a regulatory and fitness neutral function. The research identifies numerous potential candidates, either genes or regulatory areas involved in the processes that distinguish humans from other apes, such as dental and sensory perception-related characteristics. Allele sharing between current and ancient hominin genomes has been interpreted in several ways, including ancestral genetic structure or non-African introgression from archaic hominins [91]. discover 427 polymorphic human deletions that are shared with archaic hominin genomes, with roughly 87% originating before the Human-Neandertal divergence (ancient) and 9% intro gressed from Neandertals. According to the study, the genomic landscapes of both ancient and intro gressed deletion variations were predominantly sculpted by purifying selection, which eliminated big and exonic variants. The damaged genes are important in both exterior and internal chemical metabolism, growth and sperm creation, and susceptibility to psoriasis and Crohn’s disease.
Small Indels also have been discovered in most of the personal human genomes. In the Venter genome 823,396 indels were discovered, in the Watson genome 222,718 were found, and 135,262 Indels were discovered in the Han Chinese genome [92, 93, 94]. Table 3 is a list of indel discovery in human populations and personal genomes.
Coding indels have been found in several of the personal genome sequences that have been examined. For example, 739 coding Indels were identified in the Venter genome and 345 coding indels were identified in the Watson genome. Nucleotide substitution, insertion and deletion (indel) events are the major driving forces that shape genomes. Indels located in non-coding areas (introns and intergenic regions) play an important role in gene regulation. Deletions within an intronic enhancer can drastically reduce gene expression, but insertions can produce additional enhancer sites and increase expression [95]. They can also disrupt splice sites and splicing silencer/enhancer components. This can lead to incorrect splicing, leading the inclusion or exclusion of exons, which often results in mRNA degradation (nonsense-mediated decay), lowering gene product [96]. Indels can remove or change distal regulatory regions, resulting in substantial up- or down-regulation of distant genes on the chromosome [97]. They can eliminate CTCF binding sites, which are insulators that dictate 3D structure, causing a “3D genome” disruption by letting a promoter to engage inappropriately with a distal enhancer (“enhancer hijacking”) or inhibiting a necessary contact [49, 98]. Promoters contain motifs that serve as docking sites for transcription factors (TFs), which start gene transcription. Small InDels in human promoter regions are important controllers of gene expression. An indel, as small as 1-3 bp might delete or establish a new binding site, hence enhancing or decreasing the binding affinity of important TFs. For example, is the study of indels in disrupting WRKY TFs which are involved in stress responses and plant development [99]. Studies suggest that around 12% of core promoter variations (including InDels) have a significant impact on gene expression levels [49, 98]. Most of the InDels with random sequences are observed in more than one population, while approximately 28.34% [100]. One major aim of genomics research is to identify differences between genomes of species or individuals requiring assessment of genetic variation. InDels are common in different populations, in which the same alleles could be assumed to have been formed by secondary mutations at a very ancient time. In the meantime, the presence or absence of the third allele of InDels among different populations might depend on the time point of secondary mutations during diachronic population migration and expansion Parasayan, 2024.
Applications of Indel Polymorphisms
Human populations contain between 1.6 and 2.5 million indels and are found at a density of one indel per 7.2 kb [60]. Several research aims of population genetics frequently use indel data since indels are shared between global populations, however their frequency distribution differs. The study of MM-InDels reveals both consistency and differentiation among global populations [101]. African populations are the most diverse, with a greater proportion of low frequency indels (MAF < 5%). Since indels are easier to detect using high-throughput technology than other complex structural alterations they are useful in forensic identification and ancestry investigations [102]. The advantages of indels include the ability to reliably track lineage over lengthy periods of time without rapidly changes. The application in diverse genetic backgrounds and ancestry in various human populations confers an advantage in assessing the transcontinental population differences. To this end [103], discuss the use of indels in analysing transcontinental populations and ancestry inference for the Chinese Hui community. InDels are also used to assess genetic diversity and structure within and between ethnic groups or subpopulations. Subramanian’s 2024 work examine the landscape of genetic structural differences in Indian populations using indels. Because of the high polymorphism information and use of small number of markers which are sufficient for many molecular ecology and population genetics applications indels have emerged as the preferred markers. Few studies include molecular characterization and genetic diversity of Ginkgo [104]; Sesame [105], and arthropods [106]. It is extremely unlikely that two separate indel mutations of identical length will occur at the same location indicating shared organization of indels between populations. Thus, they represent identity by descent, making them extremely useful markers for reconstructing human evolutionary history [107]. Kerdoncuff [108, 109], identifies the origins of Iranian farmer-related ancestry in Indian populations and characterizes Neanderthal and Denisovan ancestry using the above premise. The use of marker loci to discover genomic areas under divergent selection is a standard method for studying local adaptation and speciation Hoban, et al., the markers themselves may be subject to direct selection or may be connected to specific loci (indirect selection). Perini et al. describe the use of indels to detect divergent selection in the intertidal snail Littorina saxatilis. Chen [110] report similar efforts in Capsicum and Chenrui [111] in Populus.
InDels are the most common type of length polymorphism and play a crucial role in the genetic features of many important phenotypes in both plants and animals, making them an excellent source of length polymorphism markers. In plants, they have been used to generate molecular maps, evaluate agronomical traits in germplasm, marker-assisted selection, and map quantitative trait loci’s (QTLs) [53], Yang describe indel markers-based map in cucumber. Trupthi, et al. present data related to agro-morphometric and molecular insights into the Western and Eastern gene pools of carrot (Daucus carota L.) [112, 113] report on the fine mapping of a QTL for plant height in soybean (Glycine max L. Merr ). In animal genomics, the marker has been used to detect disease markers, perform molecular breeding, and map QTLs [114], describe the use of indels in ewe functional genomics for detecting follicular cysts. Yang [115] describes a 14-bp indel in the PRNP gene used to analyze economic features in Chinese indigenous cattle breeds. Chen [89] reports on Indel-based QTL mapping of teat number in Qingping pigs. Advances in genome editing technologies are based on programmable nucleases (PNs) such as mega nucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR/ CRISPR-associated 9 (Cas9) nucleases. These approaches allow for genome modification at single base resolution, also possess ability to delete, insert, or replace genomic DNA in cells, organs, and complete organisms [116]. Genome editing in zygotes is an effective method for enhancing economically relevant features in cattle, as demonstrated by the knockout of the beta-carotene oxygenase (BCO2) gene linked to yellow fat disease in tan sheep [117, 118]. Jørgensen [98] report altering of the granule bound starch synthase gene GBSS in Solanum tuberosum (potato), a tetraploid organism. In cancer, the tumor suppressor gene Arhgap35 was knocked out in adult mouse liver to generate new models of liver cancer Alves-Bezerra et al., 2019. Adaptive cancer immunotherapy is a therapeutic application that involves knocking down the immunoregulatory gene PDCD1 in patient-derived tumor infiltrating lymphocytes (TILs) to boost the anti-tumor activity of the T-cell population Chamberlain. In human genomics, indels have been utilized to determine fetal sex with cell-free fetal DNA from maternal plasma [119]. They are employed in association studies of numerous Mendelian and complex traits such as skeletal muscle hypertrophy [120, 121], ocular disorders [122, 123, 124] and Zhang [125]. Indels are critical in the diagnosis, risk assessment, and prognosis of a variety of human diseases, including neurological disorders, cancer, and cardiovascular disease. In Cancer diagnostics and prognosis indels in IL4, TYMS, UCP2, and HLA-G genes are linked to colorectal cancer risk, with specific alleles indicating higher tumor-node- metastasis (TNM) stages or increased recurrence risk [126]. The ACE gene InDel has been identified as a susceptibility marker for Alzheimer’s disease [127]. Further, InDels in MIR155HG and CTH genes have been discovered as risk factors in cardiovascular disease and sudden cardiac death (SCD) [128]. The ACE gene I/D polymorphism is associated with COVID-19 severity, correlating with increased risk of severe infection and hospitalization. Alaa [129] Indels
acts as reliable anchors for tracking recombination events within highly repetitive or variable genomic regions, such as the P. falciparum pfmdr1 locus. Miles [130] report several instances of meiotic recombination within copy number variants associated with drug resistance in P. falciparum. Chromosomal rearrangements such as indels or duplications, can occur by transposition, unequal homologous recombination, or illegitimate recombination. Wang [131] report changes in non-homologous end-joining mediated gene changes in lymphocyte development. Table-4 is a representative list of genes with possible mechanism and path of Insertion/Deletion events.
Due to small amplicon sizes (~100 bp), they are highly effective in testing degraded DNA samples, such as skeletal material or paraffin-embedded tissues. Since the typing of indels is cost-effective, they serve as excellent molecular markers in wildlife biology offering high accuracy, co- dominant inheritance, and ease of analysis, especially for non-model organisms and degraded DNA samples offering a non-invasive typing of DNA (e.g., hair, scat, feathers) Zupanič Pajnič, et al. They enable quantification of genetic variation within a population, helping to detect inbreeding or population bottlenecks (e.g., studies on endangered mammals). They also aid in the differentiation of isolated populations, allowing suitable populations to be translocated in order to boost genetic diversity. Few applications of indels include study of long-term mixing of grey wolves and domestic dogs across [132]. Parker used scat samples to identify, sex determination, measurement of genetic diversity, and effective population sizes in coyotes and kit foxes. Finally, Pilot [133], describe the use of indels in koala populations to improve reproductive success and prevent inbreeding.
Discussion
With the human genome sequencing projects successfully completed, the next task for biologists is to decipher the instructions encoded in the genomes. Junk DNA, also known as “non-coding DNA,” refers to sequences in the genome that do not encode proteins and do not appear to have a distinct, vital role for the organism [134]. While originally thought to be completely non-coding, it is now realized that this DNA (which accounts for around 90-98% of the human genome) can play structural roles, act as regulatory elements, or also serve as evolutionary baggage. Junk DNA contains pseudogenes (dead gene copies), introns (non-coding sections inside genes), and a large number of repetitive sequences, such as indels, transposons and retrotransposons [135]. Short indels are the second most common type of human genetic variation, accounting for a significant portion of genetic variation in humans and influencing a wide range of human phenotypes. The recent availability of low-cost, high-throughput sequencing has enabled the cataloging of new indels in individual genomes from numerous mammals and other species [136]. An often-asked question in biology is why “junk DNA” is not removed from the original genome after millions of years of evolution? DNA lengths can be removed or inserted into a gene and genes or gene segments can be inverted or duplicated. Studies implicate that indels, rather than substitutions, account for the majority of genomic divergence. Therefore, the study of the patterns of insertion and deletion is necessary to understand the evolution of mammalian genomes.
With reference to the organization, structure and replication of indel DNA complexity analysis has enabled to examine the possible intermediates through which indel could have occurred and to propose likely mechanisms and pathways for indel generation [137]. The current accepted mechanisms include a two-step insertion/deletion process assessed in the context of 10 base-pairs DNA sequence flanking the lesion on either side. The postulated mechanisms underlying micro-deletions and micro-insertions serve as templates for templates for indel replication. Recent research in this area of the indel include isogenic CRISPR-edited human cellular models of post-replicative repair dysfunction (PRRd), including individual and combined gene edits of DNA mismatch repair (MMR) and replicative polymerases (Pol ε and Pol δ). The study reveals unique, diverse indel mutational footprints [138]. Accurate Indel characterization is crucial for biological and clinical purposes. High proportions of microhomology-mediated deletion are now utilized as key predictor of clinically actionable homologous recombination deficiency in several human disease. The change in complexity consequent to a mutation determines the type of repeat sequence involved in mediating the event, thereby providing clues as to the underlying mutational mechanism.
This class of indels can help address various questions in genetics-meiotic and recombination biology, including the significance of DNA organization and stoichiometry. Also, Indels influence numerous elements of recombination and related genetic mechanisms, resulting in reduced genetic exchange, crossover activity, and topological limitations [130]. They activate the error-prone DNA repair mechanism, causing nucleotide alterations at the mutation site. Indels are frequently used as mutation substrates because of their disruptive effect on gene conversion. Thus, indels affect meiotic and recombination in several. However, Indels, particularly those involving AT-rich regions, enable studies on repair processes during recombination favoring GC over AT, also detect meiotic drive and inheritance of specific alleles from parental genotype. According to studies, deletions are subjected to increased purifying selection inside genes and across the genome, with structural position and flanking sequence serving as decisive variables. The high selection restriction against indel mutations implies that indels may contribute to human phenotypic variation and GWAS and eQTL data.
Indel is a unique evolutionary event that serves as an effective indicator of shared ancestry and evolutionary branching points. Several studies in evolutionary biology have used this advantage to identify lineages of various plants and animals. The current availability of primate genomes has enabled major applications of indel in resolving primate phylogenetic relationships, identifying human-specific (HS) lineage alterations, and admixture analysis [139, 140]. The rate at which indel “Molecular Clocks” accumulate in non-coding areas allows for the estimation of divergence time between closely related populations. Thus, they are effective markers for a variety of lineage analyses, providing a “slower” but more stable evolutionary signal. The research efforts in human evolution have helped identification of several unique human genes involved in metabolism, growth and sperm production, and illness susceptibility.
The study of indel provides various opportunities to investigate the role of repetitive DNA in genome organization, regulation, and pathogenesis [141]. Indel mutation rates are highly variable throughout the genome. Indel frequencies are also dramatically reduced throughout a wide range of functional noncoding regions in the human genome, with large reductions observed in UTRs and introns. This is demonstrated by the clustering of indels within homopolymer run (HR), Tandem repeats (TR), and PR sites (hotspots), which account for 43%-48% of called indels while occupying only 4.03% of the genome sequence [142]. Higher mutation rates in HR and TR sites raise questions about their evolutionary dynamics, as to whether such sites develop or diminish over time. Among polarized indels, deletions exceed insertions, implying the presence of a metastable equilibrium between HR and TR tracts. An initial screen revealed 43 genes with high individual projected mutation rates within the coding regions, with frame-preserving indels measuring 3 bp in length being the most common [143]. The presence of indels in both the coding and non-coding regions of the gene has an impact on transcription and gene regulation, underscoring their importance in the genome [144].
According to population genetics studies, InDels are shared globally, but their frequency distributions vary by population. Because they can track lineage over long periods of time without alteration, they are widely employed in forensics, ancestry, and many molecular ecological investigations. Since indels are abundant and distributed throughout the genomes they serve as excellent source of length polymorphisms, play critical roles in the genetic characteristics of many essential traits in both plants and animals. The selective advantage of these markers has made inroads into wildlife biology research to answer a variety of questions. The application of these markers in the field of biomedical sciences as reliable and easily genotyped markers includes association with various human disorders, for prognosis and indicators, and disease grade assessment. Next-generation sequencing technologies have ushered in a new era of human genome sequencing. Human genomes are being sequenced at unprecedented rates, and the era of tailored treatment is clinics globally now practiced [145]. These advancements will enable increased application of indel in human disease biology.
Ohno’s original idea on the origin and evolutionary importance of “junk DNA” has been changed and refined over the previous four decades [146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170], aided by modern comparative and functional genomics research in several species, as well as large-scale bioinformatics sequence analysis. Advances in the research of indel polymorphism are one such example adding small “nugget” to the functional roles of “junk DNA”. In future it is assumed that the variation profiles will also be used to predict a variety of other human traits such as height, weight, appearance, longevity, and intellectual quotient (IQ), a area of human genetics where indels will be the markers of choice [171, 172, 173, 174, 175, 176, 177, 178, 179, 180, 181, 182, 183, 184, 185, 186, 187, 188, 189].
Conclusion
Several million indels have been identified in human populations and global personal genomes thanks to the global efforts in sequencing. These small indels cause a large amount of genomic variation, affect various aspects of recombination and are major elements in molecular evolution. Further, these markers have been frequently used as tools in several areas of biology, including DNA structure- function correlations, genome organization, and genome stability. With the availability of modern high-throughput and calling bioinformatics technologies, it is envisaged that biology researchers will be able to better use the indel polymorphisms to address wide range of topics.
References
-
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB (2010) Annotating non-coding regions of the genome. Nat Rev Genet 11(8): 559-571.
-
Liao X, Zhu W, Zhou J, Li H, Xu X (2023) Repetitive DNA sequence detection and its role in the human genome. Communications biology 6(1): 954.
-
Šatović-Vukšić E, Majcen P, Plohl M (2025) Satellite DNAs rising from the transposon graveyards. Dna research 32(5): dsaf026.
-
Šatović-Vukšić E, Plohl M (2021) Classification Problems of Repetitive DNA Sequences. DNA 1(2): 84-90.
-
Jurka J, Bao W, Kojima K, Kapitonov VV (2011) Repetitive elements: bioinformatic identification, classification and analysis.
-
Amiteye S (2021) Basic concepts and methodologies of DNA marker systems in plant molecular breeding. Heliyon 7(10): e08093.
-
Brown TA (2020) Gene Cloning and DNA Analysis: An Introduction. In: 8th (Edn.), Wiley-Blackwell. USA, pp: 432.
-
Challis D, Antunes L, Garrison E, Quan M, Chen B, et al. (2015) The distribution and mutagenesis of short coding INDELs from 1,128 whole exomes. BMC Genomics 16(1): 143.
-
Qi M, Stenson PD, Ball EV, Tainer JA, Bacolla A, et al. (2022) Distinct sequence features underlie microdeletions and gross deletions in the human genome. Hum Mutat 43(3): 328-346.
-
Hollister JD, Ross Ibarra J, Gaut BS (2010) Indel- associated mutation rate varies with mating system in flowering plants. Mol Biol Evol 27(2): 409-416.
-
Cooper DN, Bacolla A, Férec C, Vasquez KM, Kehrer- Sawatzki H (2011) On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat 32(10): 1075-1099.
-
Mehta A, Haber JE (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6(9): a016428.
-
Stinson BM, Loparo JJ (2021) Repair of DNA Double- Strand Breaks by the Nonhomologous End Joining Pathway. Annu Rev Biochem 90: 137-164.
-
Bakhache W, Symonds-OrrW, McCormick L, Dolan PT (2025) Deep mutation, insertion and deletion scanning across the Enterovirus A proteome reveals constraints shaping viral evolution. Nat Microbiol 10: 158-168.
-
Casaubon JT, Kashyap S, Regan JP (2025) BRCA1 and BRCA2 Mutations. StatPearls Publishing, Treasure Island (FL).
-
Dai J, Huang M, Amos CI, Hung RJ, Tardon A, et al. (2020) Genome-wide association study of INDELs identified four novel susceptibility loci associated with lung cancer risk. Int J Cancer 146: 2855-2864.
-
Dittwald P, Gambin T, Gonzaga-Jauregui C, Carvalho CM, Lupski JR (2013) Inverted low-copy repeats and genome instability-a genome-wide analysis. Hum Mutat 34(1): 210-220.
-
Achar A, Sætrom P (2015) RNA motif discovery: a computational overview. Biol Direct 10: 61.
-
Chen FC, Chen CJ, Li WH, Chuang TJ (2007) Human- specific insertions and deletions inferred from mammalian genome sequences. Genome Res 17: 16-22.
-
Hu J, Ng PC (2013) SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS One 8: e77940.
-
Mehrotra S, Goyal V (2014) Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genomics Proteomics Bioinformatics 12(4): 164-171.
-
Polleys EJ, Freudenreich CH (2021) Homologous recombination within repetitive DNA. Curr Opin Genet Dev 71: 143-153
-
Saada AA, Lambert SAE, Carr AM (2018) Preserving replication fork integrity and competence via the homologous recombination pathway. DNA repair 71: 135-147.
-
Brown RE, Freudenreich CH (2021) Structure-forming repeats and their impact on genome stability. Curr Opin Genet Dev 67: 41-51.
-
McGinty RJ, Balick DJ, Mirkin SM, Sunyaev SR (2025) Inherent instability of simple DNA repeats shapes an evolutionarily stable distribution of repeat lengths. Nature Communications 17: 93.
-
Kaushal S, Freudenreich CH (2019) The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 58(5): 270-283.
-
Casas-Delucchi CS, Daza-Martin M, Williams SL, Coster G (2022) The mechanism of replication stalling and recovery within repetitive DNA. Nat Commun 13(1): 3953.
-
Neil AJ, Kim JC, Mirkin SM (2017) Precarious maintenance of simple DNA repeats in eukaryotes. BioEssays 39: 1-10.
-
Richard GF, Paques F (2000) Mini- and microsatellite expansions: the recombination connection. EMBO Rep 1: 122-126.
-
Vervoort L, Vermeesch JR (2023) Low copy repeats in the genome: from neglected to respected. Explor Med 4: 166-175.
-
Reid DA, Conlin MP, Yin Y, Chang HH, Watanabe G, et al. (2017) Bridging of double-stranded breaks by the nonhomologous end-joining ligation complex is modulated by DNA end chemistry. Nucleic Acids Research 45(4): 1872-1878.
-
Piazza A, Heyer WD (2019) Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol 29(2): 135-149.
-
Vítor AC, Huertas P, Legube G, de Almeida SF (2020) Studying DNA Double-Strand Break Repair: An Ever- Growing Toolbox. Front Mol Biosci 7: 24.
-
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, et al. (2011) Dindel: Accurate indel calls from short- read data. Bioinformatics 27(15): 2047-2054.
-
Narzisi G, Schatz MC (2015) The challenge of small-scale repeats for indel discovery. Bioinformatics 31(8): 1263- 1269.
-
Qin D (2019) Next-generation sequencing and its clinical application. Cancer Biol Med 16(1): 4-10.
-
Li H, Homer N (2010) A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics 11(5): 473-483.
-
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
-
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z (2009) Pindel: A pattern growth approach to detect breakpoints of large deletions and medium-sized insertions. Bioinformatics 25(21): 2865-2871.
-
Narzisi G, O Rawe JA, Iossifov I, Fang H, Lee YH, et al. (2014) Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nature Methods 11(10): 1033-1036.
-
Logsdon GA, Vollger MR, Eichler EE (2020) Long-read human genome sequencing and its applications. Nature Reviews Genetics 21(10): 597-614.
-
1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526(7571): 68-74.
-
Boatwright JL, Sapkota S, Kresovich S (2023) Functional genomic effects of indels using Bayesian genome- phenome wide association studies in sorghum. Front Genet 14: 1143395.
-
Barbosa IG, Ferreira GC, Andrade DF, Januário CR, Belisário AR, et al. (2020) The renin angiotensin system and bipolar disorder: a systematic review. Protein and Peptide Letters 27(6): 520-528.
-
Zhang D, Cheng L, Qian Y, Alliey-Rodriguez N, Kelsoe JR, et al. (2009) Singleton deletions throughout the genome increase risk of bipolar disorder. Molecular psychiatry 14(4): 376-380.
-
Wongpaiboonwattana W, Hnoonual A, Limprasert P (2022) Association between 19-bp insertion/deletion polymorphism of dopamine β-hydroxylase and autism spectrum disorder in Thai patients. Medicina 58(9): 1228.
-
Shiina A, Ishikawa D, Ishizawa K, Kasahara H, Fujita Y, et al. (2024) Alexander disease with a novel GFAP insertion- deletion mutation mimicking progressive supranuclear palsy. Clin Neurol Neurosurg 240: 108261.
-
Lin BT, Chien CF, Huang LC, Yang YH (2025) Association Between Angiotensin‐Converting Enzyme (ACE) Gene Insertion/Deletion (I/D) Polymorphism Genotypes with Brain Volume and Hypertension in Alzheimer’s Disease—A Retrospective Study. Kaohsiung J Med Sci 41(9): e70046.
-
Liu Y, Wang J, Liu S, Kuang M, Jing G, et al. (2019) A novel transgenic murine model with persistently brittle bones simulating osteogenesis imperfecta type I. Bone 127: 646-655.
-
Yang PJ, Ting KH, Tsai PY, Su SC, Yang SF (2024) Association of long noncoding RNA GAS5 gene polymorphism with progression of diabetic kidney disease. International Journal of Medical Sciences 21(11): 2201.
-
Yuda A, Nakamura T, Momose S, Ishii S, Tanaka H, et al. (2025) A comprehensive approach for identifying filaggrin mutations and copy number variants by long- read sequencing. Genomics 117(4): 111055.
-
Temel SG, Ergoren MC, Yilmaz I, Oral HB (2019) The use of ACE INDEL polymorphism as a biomarker of coronary artery disease (CAD) in humans with Mediterranean- style diet. Int J Biol Macromol 123: 576-580.
-
Yang J, Meng P, Mi H, Wang X, Yang J, et al. (2025) The development of ideal insertion and deletion (InDel) markers and initial indel map variation in cucumber using re-sequenced data. BMC Genomics 26(1): 391.
-
Dong Y, Xue R, Zhang Y, Jia X, Jiang M, et al. (2026) Fundus albipunctatus disease-associated RDH5/ L310delinsEV mutation undertakes AMFR-mediated polyubiquitination and degradation in proteasome. Experimental Eye Research 110927.
-
Tao R, Hu S, Wang S, Zhou X, Zhang Q, et al. (2015) Association between indel polymorphism in the promoter region of lncRNA GAS5 and the risk of hepatocellular carcinoma. Carcinogenesis 36(10): 1136-1143.
-
Guo H, Han Y, Zhou Q, Chen J, Wang M, et al. (2024) GABPα inhibits tumor progression and angiogenesis via a novel 18-bp indel within VEGF promoter in breast cancer. Cancer Biomarkers 41(3-4): CBM-230541.
-
Ferrer-Torres P, Galván-Femenía I, Supek F (2025) Joint inference of mutational signatures from indels and single-nucleotide substitutions reveals prognostic impact of DNA repair deficiencies. Genome Medicine 17(1): 76.
-
Kvikstad EM, Chiaromonte F, Makova KD (2009) Ride the wavelet: A multiscale analysis of genomic contexts flanking small insertions and deletions. Genome Res 19(7): 1153-64.
-
Ziolkowski PA, Henderson IR (2017) Interconnections between meiotic recombination and sequence polymorphism in plant genomes. New Phytol 213: 1022- 1029.
-
Porubsky D, Hops W, Ashraf H, Hsieh PH, Rodriguez-Martin B, et al. (2022) Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 185(11): 1986-2005.e26.
-
Berdan EL, Blanckaert A, Butlin RK, Bank C (2021) Deleterious mutation accumulation and the long-term fate of chromosomal inversions. PLoS Genet 17(3): e1009411.
-
Shivaprasad KM, Aski M, Mishra GP, Sinha SK, Gupta S, et al. (2024) Genome-wide discovery of InDels and validation of PCR-Based InDel markers for earliness in a RIL population and genotypes of lentil (Lens culinaris Medik.). PLoS ONE 19(5): e0302870.
-
Coelho MC, Pinto RM, Murray AW (2019) Heterozygous mutations cause genetic instability in a yeast model of cancer evolution. Nature 566(7743): 275-278.
-
Biba D, Klink G, Bazykin GA (2022) Pairs of Mutually Compensatory Frameshifting Mutations Contribute to Protein Evolution. Mol Biol Evol 39(3): msac031.
-
Kalmari A, Colagar AH, Heydari M, Arash V (2023) Missense polymorphisms potentially involved in mandibular prognathism. J Oral Biol Craniofac Res 13(3): 453-460.
-
Martoni E, Petrini S, Trabanelli C, Sabatelli P, Urciuolo A, et al. (2013) Characterization of a rare case of Ullrich congenital muscular dystrophy due to truncating mutations within the COL6A1 gene C-Terminal domain: a case report. BMC Med Genet 14: 59.
-
Zhang X, Lin H, Zhao H, Hao Y, Mort M, et al (2014) Impact of human pathogenic micro-insertions and micro-deletions on post-transcriptional regulation. Hum Mol Genet 23(11): 3024-3034.
-
Holm IA, Agrawal PB, Ceyhan-Birsoy O, Christensen KD, Fayer S (2018) The BabySeq project: implementing genomic sequencing in newborns. BMC Pediatr 18(1): 225
-
Lalonde S, Stone OA, Lessard S, Lavertu A, Desjardins J, et al. (2017) Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS One 12(6): e0178700.
-
Chen J, Guo JT (2021) Structural and functional analysis of somatic coding and UTR indels in breast and lung cancer genomes. Sci Rep 11: 21178.
-
Zhang R, Yan H, Tian F, Jiang Y, Chen Y, et al. (2025) Prognostic implications of uncommon EGFR exon 19 deletion-insertion mutations in non-small cell lung cancer treated with third-generation EGFR-TKIs. Lung Cancer 108755.
-
Marand AP, Chen Z, Gallavotti A, Schmitz RJ (2021) A cis- regulatory atlas in maize at single-cell resolution. Cell 184(112021): 3041-3055.
-
Horton JS, Flanagan LM, Jackson RW, Priest NK, Taylor TB (2021) A mutational hotspot that determines highly repeatable evolution can be built and broken by silent genetic changes. Nat Commun 12(1): 6092
-
Moutinho AF, Bataillon T, Dutheil JY (2020) Variation of the adaptive substitution rate between species and within genomes. Evol Ecol 34(3): 315-338.
-
Barton HJ, Zeng K (2018) New Methods for Inferring the Distribution of Fitness Effects for INDELs and SNPs. Mol Biol Evol 35(6): 1536-1546.
-
Cvijović I, Good BH, Desai MM (2018) The Effect of Strong Purifying Selection on Genetic Diversity. Genetics 209(4): 1235-1278.
-
Dussex N, Morales HE, Grossen C, Dalén L, van Oosterhout C (2023) Purging and accumulation of genetic load in conservation. Trends in Ecology & Evolution 38(10): 961-969.
-
Corcoran P, Gossmann TI, Barton HJ (2017) The Great Tit HapMap Consortium, Jon Slate, Kai Zeng, Determinants of the Efficacy of Natural Selection on Coding and Noncoding Variability in Two Passerine Species. Genome Biol Evol 9(11): 2987-3007.
-
Samano A, Musat M, Junaghare M, Ahmad A, Ali M, et al. (2025) Structural variants are enriched in deleterious visible phenotypes in Drosophila. BioRxiv 15.670616
-
McComish BJ, Charleston MA, Parks M, Baroni C, Salvatore MC, et al. (2024) Ancient and Modern Genomes Reveal Microsatellites Maintain a Dynamic Equilibrium Through Deep Time. Genome Biol Evol 16(3): evae017.
-
Young RS (2016) Lineage-specific genomics: Frequent birth and death in the human genome: The human genome contains many lineage-specific elements created by both sequence and functional turnover. Bioessays 38(7): 654-63.
-
Hubert KA, Wellik DM (2023) Hox genes in development and beyond. Development 150(1): dev192476.
-
Kashfi SM, Farahbakhsh FB, Golmohammadi M, Mojarad NE, Azimzadeh P, et al. (2014) Frameshift Mutations (Deletion at Codon 1309 and Codon 849) in the APC Gene in Iranian FAP Patients: a Case Series and Review of the Literature. Int J Mol Cell Med Summer 3(3): 196-202.
-
Elena SF (2023) The role of indels in evolution and pathogenicity of RNA viruses. Proc Natl Acad Sci U S A 120(33): e2310785120.
-
Rangel MA, Dolan PT, Taguwa S, Xiao Y, Andino R, et al. (2023) High-resolution mapping reveals the mechanism and contribution of genome insertions and deletions to RNA virus evolution. Proc Natl Acad Sci U S A 120 (31): e2304667120.
-
Paśko L, Ericson PGP, Elzanowski A (2011) Phylogenetic utility and evolution of indels: A study in neognathous birds. Molecular Phylogenetics and Evolution 61(3): 760-771.
-
Hu J, Ng PC (2012) Predicting the effects of frameshifting indels. Genome Biol 13: R9.
-
Biller P (2025) Evolution of ultraconserved elements by indels. BioRxiv 27: 656252.
-
Chen M, Yang C, Zhai X, Wang C, Liu M, et al. (2024) Comprehensive Identification and Characterization of HML-9 Group in Chimpanzee Genome. Viruses 16(6): 892.
-
Elizabeth HBH, Kern AD (2015) The Role of DNA Insertions in Phenotypic Differentiation between Humans and Other Primates. Genome Biology and Evolution 7: 1168-1178.
-
Witt KE, Villanea F, Loughran E, Zhang X, Huerta-Sanchez E (2022) Apportioning archaic variants among modern populations. Philos Trans R Soc Lond B Biol Sci 377 (1852): 20200411.
-
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, et al. (2007) The diploid genome sequence of an individual human. PLoS Biol 5.
-
Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, et al. (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature, 452: 872- 876.
-
Wang J, Wang W, Li R, Li Y, Tian G, et al. (2008) The diploid genome sequence of an Asian individual. Nature 456: 60-65.
-
Meng F, Zhao H, Zhu B, Zhang T, Yang M, et al. (2021) Genomic editing of intronic enhancers unveils their role in fine-tuning tissue-specific gene expression in Arabidopsis thaliana. Plant Cell 33(6): 1997-2014.
-
Anna A, Monika G (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet 59: 253-268.
-
Yao L, Berman BP, Farnham PJ (2015) Demystifying the secret mission of enhancers: linking distal regulatory elements to target genes. Crit Rev Biochem Mol Biol 50(6): 550-573.
-
Jørgensen B, Liu Y, Bennett EP, Andreasson E, Nielsen KL, et al. (2019) High efficacy full allelic CRISPR/Cas9 gene editing in tetraploid potato. Sci Rep 9: 17715.
-
Li Y, Li Z, Xu C, Wang Q (2025) WRKYs as regulatory hubs of secondary metabolic networks: Diverse inducers and distinct responses. Plant Communications 6(9): 101438.
-
Prakrithi P, Singhal K, Sharma D, Jain A, Bhoyar RC, et al. (2022) An Alu insertion map of the Indian population: identification and analysis in 1021 genomes of the IndiGen project. NAR Genomics and Bioinformatics 4(1): lqac009.
-
Yao Y, Sun K, Yang Q, Zhou Z, Shao C (2022) Assessing Autosomal InDel Loci With Multiple Insertions or Deletions of Random DNA Sequences in Human Genome. Front Genet 12: 809815.
-
Du W, Peng Z, Feng C, Zhu B, Wang B (2017) Forensic efficiency and genetic variation of 30 InDels in Vietnamese and Nigerian populations. Oncotarget 8(51): 88934-88940.
-
Xie T, Shen C, Jin X, Lan Q, Fang Y, et al. (2020) Genetic Structural Differentiation Analyses of Intercontinental Populations and Ancestry Inference of the Chinese Hui Group Based on a Novel Developed Autosomal AIM-InDel Genotyping System. Biomed Res Int 2020: 2124370.
-
Wang D, Zhou Q, Le L, Fu F, Wang G (2023) Molecular Characterization and Genetic Diversity of Ginkgo (Ginkgo biloba L.) Based on Insertions and Deletions (InDel) Markers. Plants 12(13): 256.
-
Kizil S, Basak M, Guden B, Tosun HS, Uzun B (2020) Genome-Wide Discovery of InDel Markers in Sesame (Sesamum indicum L.) Using ddRADSeq. Plants 9(10): 1262.
-
Emerson BC, Borges PAV, Cardoso P, Convey P, deWaard JR, et al. (2023) Collective and harmonized high throughput barcoding of insular arthropod biodiversity: Toward a Genomic Observatories Network for islands. Molecular Ecology 32: 6161-6176.
-
Glodzik D, Navarro P, Vitart V, Hayward C, McQuillan R, et al. (2013) Inference of identity by descent in population isolates and optimal sequencing studies. Eur J Hum Genet 21(10): 1140-1145.
-
Kerdoncuff E, Skov L, Patterson N, Banerjee J, Khobragade P, et al. (2025) Exploring functional InDels and genetic diversity: agro-morphometric and molecular insights into the Western and Eastern gene pools of carrot (Daucus carota L.). Frontiers in Plant Science 16.
-
Kerdoncuff E, Laurits S, Patterson N, Joyita B, Pranali K, et al. (2025) 50,000 years of evolutionary history of India: Impact on health and disease variation. Cell 188(13): 3389-3404.
-
Chen J, Guo JT (2020) Comparative assessments of indel annotations in healthy and cancer genomes with next-generation sequencing data. BMC Med Genomics 13(1): 170.
-
Chenrui G, Qingzhang D, Jianbo X, Mingyang Q, Beibei C, et al. (2017) Dissection of Insertion-Deletion Variants within Differentially Expressed Genes Involved in Wood Formation in Populus. Frontiers in Plant Science 8.
-
Liu XS, Wu H, Krzisch M, Wu X, Graef J, et al. (2018) Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172(5): 979- 992.
-
Liu Z, Li H, Zhong Z, Jiang S (2022) A Whole Genome Sequencing-Based Genome-Wide Association Study Reveals the Potential Associations of Teat Number in Qingping Pigs. Animals 12(9): 1057.
-
Wang H, Liu C, Zhang T, Sa Q, Li B (2025) InDels of ewe functional genes and their association with follicular cysts. Small Ruminant Research 251: 107569.
-
Yang Q, Zhang S, Liu L, Cao X, Lei C (2016) Application of mathematical expectation (ME) strategy for detecting low frequency mutations: An example for evaluating 14- bp insertion/deletion (indel) within the bovine PRNP gene. Prion 10(5): 409-419.
-
Gaj T, Sirk SJ, Shui SL, Liu J (2016) Genome-Editing Technologies: Principles and Applications. Cold Spring Harb Perspect Biol 8(12): a023754.
-
Guo C, Du J, Wang L, Yang S, Mauricio R, et al. (2016) Insertions/Deletions-Associated Nucleotide Polymorphism in Arabidopsis thaliana. Front Plant Sci 7: 1792.
-
Wang X, Niu Y, Zhou J, Yu H, Kou Q, et al. (2016) Multiplex gene editing via CRISPR/Cas9 exhibits desirable muscle hypertrophy without detectable off- target effects in sheep. Sci Rep 6: 32271.
-
Ho SS, Barrett A, Thadani H, Asibal CL, Koay ES, et al. (2015) Application of real-time PCR of sex-independent insertion-deletion polymorphisms to determine fetal sex using cell-free fetal DNA from maternal plasma. Clin Chem Lab Med 53(8): 1189-1195.
-
Kazan HH, Kasakolu A, Koncagul S, Ergun MA, John G, et al. (2025) Association analysis of indel variants and gene expression identifies MDM4 as a novel locus for skeletal muscle hypertrophy and power athlete status. Experimental Physiology 110: 1661-1671.
-
Marques D, Ferreira-Costa LR, Ferreira-Costa LL, Correa RDS, Borges AMP, et al. (2017) Association of insertion-deletions polymorphisms with colorectal cancer risk and clinical features. World J Gastroenterol 23(37): 6854-6867.
-
Singh M, Tyagi SC (2018) Genes and genetics in eye diseases: a genomic medicine approach for investigating hereditary and inflammatory ocular disorders. Int J Ophthalmol 11(1): 117-134.
-
Krynytska I, Kucher S, Tokarskyy O, Koval M, Marushchak M (2021) The association of angiotensin- converting enzyme gene insertion/deletion polymorphism with bronchial asthma. Pol Merkur Lekarski 49(294): 442-444.
-
Bordbar M, Saadat M (2023) Association between 15 insertion/deletion genetic polymorphisms and risk of schizophrenia using pooled samples. EXCLI J 22: 310- 314.
-
Shah M, Gupta A, Talekar M, Chaaithanya K, Doctor P, et al. (2024) The ‘Insertion/Deletion’ Polymorphism, rs4340 and Diabetes Risk: A Pilot Study from a Hospital Cohort. Indian J Clin Biochem 39(1): 124-129.
-
Zhang Y, He S, Yu L, Shi C, Zhang Y, et al. (2023) Prognostic significance of HLA-G in patients with colorectal cancer: a meta-analysis and bioinformatics analysis. BMC Cancer 23(1): 1024.
-
Xin XY, Lai ZH, Ding KQ, Zeng LL, Ma JF (2021) Angiotensin-converting enzyme polymorphisms AND Alzheimer’s disease susceptibility: An updated meta- analysis. PLoS One 16(11): e0260498.
-
Zhang Q, Yu H, Yang Z, Li L, He Y, et al. (2021) A Functional Indel Polymorphism Within MIR155HG Is Associated with Sudden Cardiac Death Risk in a Chinese Population. Front Cardiovasc Med 8: 671168.
-
Alaa A, Sarhan N, Lotfy El-Ansary MG, Bazan NS, Farouk K, et al. (2023) Association between genetic polymorphism, severity, and treatment response among COVID-19 infected Egyptian patients. Front Pharmacol 14: 1209286.
-
Miles A, Iqbal Z, Vauterin P, Pearson R, Campino S, et al. (2016) Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res 26(9): 1288-1299.
-
Wang XS, Lee BJ, Zha S (2020) The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair 94: 102874.
-
Pilot M, Greco C, vonHoldt BM, Randi E, Jędrzejewski W, et al. (2018) Widespread, long-term admixture between grey wolves and domestic dogs across Eurasia and its implications for the conservation status of hybrids. Evol Appl 11(5): 662-680
-
Schultz AJ, Strickland K, Cristescu RH, Hanger J, de Villiers D, et al. (2021) Testing the effectiveness of genetic monitoring using genetic non-invasive sampling. Ecol Evol 12(1): e8459.
-
Pagni S, Mills JD, Frankish A, Mudge JM, Sisodiya SM (2022) Non-coding regulatory elements: Potential roles in disease and the case of epilepsy. Neuropathol Appl Neurobiol 48(3): e12775.
-
Shanmugam A, Nagarajan A, Pramanayagam S (2017) Non-coding DNA - a brief review. Journal of Applied Biology & Biotechnology 5(05): 42-47.
-
Satam H, Joshi K, Mangrolia U, Waghoo S, Zaidi G, et al. (2023) Next-Generation Sequencing Technology: Current Trends and Advancements. Biology (Basel) 12(7): 997.
-
Boschiero C, Gheyas AA, Ralph HK (2015) Detection and characterization of small insertion and deletion genetic variants in modern layer chicken genomes. BMC Genomics 16: 562.
-
Koh GCC, Nanda AS, Rinaldi G, Boushaki S, Degasperi A, et al. (2025) A redefined InDel taxonomy provides insights into mutational signatures. Nat Genet 57: 1132- 1141.
-
Mao Y, Harvey WT, Porubsky D, Munson KM, Hoekzema K, et al. (2024) Structurally divergent and recurrently mutated regions of primate genomes. Cell 187(6): 1547-1562.
-
Tan X, Qi J, Liu Z, Fan P, Liu G, et al. (2023) Phylogenomics Reveals High Levels of Incomplete Lineage Sorting at the Ancestral Nodes of the Macaque Radiation. Molecular Biology and Evolution 40(11): msad229.
-
Dunn MJ, Anderson MZ (2019) To Repeat or Not to Repeat: Repetitive Sequences Regulate Genome Stability in Candida albicans. Genes 10(11): 866.
-
Redelings BD, Holmes I, Lunter G, Pupko T, Anisimova M (2024) Insertions and Deletions: Computational Methods, Evolutionary Dynamics, and Biological Applications. Mol Biol Evol. 41(9): msae177.
-
Deng S, Song H, Li C (2025) A deep learning framework for building INDEL 2 mutation rate maps 3. BioRxiv 18: 689146.
-
Dhaene E, Vergult S (2021) Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genetics in Medicine 23(1): 34-46
-
Brlek P, Bulić L, Bračić M, Projić P, Škaro V, et al. (2024) Implementing Whole Genome Sequencing (WGS) in Clinical Practice: Advantages, Challenges, and Future Perspectives. Cells 13(6): 504.
-
Fagundes NJR, Bisso-Machado R, Figueiredo PICC, Varal M, Zani ALS (2022) What We Talk About When We Talk About “Junk DNA”. Genome Biol Evol 14(5): evac055.
-
Aleksandre J, Yang W, Dekker C, Nasser W, Muskhelishvili G (2021) DNA sequence-directed cooperation between nucleoid-associated proteins. iScience 24(5): 102408.
-
Azhaguraja M, Sankaralingam S, Anitha P, Binoj C, Aravindakshan TV (2024) Characterization of 24bp Insertion Polymorphism of Prolactin Gene and its Association with Quantitative Traits in Tellicherry Native Chicken Breed. Indian Journal of Animal Research 58(3): 376-380.
-
Barnabé C, Brenière SF, Santillán-Guayasamín S, Douzery EJ, Waleckx E (2023) Revisiting gene typing and phylogeny of Trypanosoma cruzi reference strains: Comparison of the relevance of mitochondrial DNA, single-copy nuclear DNA, and the intergenic region of mini-exon gene. Infection, Genetics and Evolution 115: 105504.
-
Barton HJ, Zeng K (2019) The Impact of Natural Selection on Short Insertion and Deletion Variation in the Great Tit Genome. Genome Biol Evol 11(6): 1514- 1524.
-
Benedetto CD, Tsark A, Acenas D, Thach A, Singhal A, et al. (2025) An InDel Genomic Variant within a Bifunctional Super-Enhancer for LINC00636 and CD47 Regulation in Breast Cancer. bioRxiv 11.
-
Bhoge Gowda KKH (2025) Repetitive DNA and Its Roles in Diverse Facets of Biology. pp: 1-118.
-
Casas-Delucchi CS, Daza-Martin M, Williams SL, Coster G (2022) The mechanism of replication stalling and recovery within repetitive DNA. Nat Commun 13(1): 3953.
-
Chintalapati M, Dannemann M, Prüfer K (2017) Using the Neandertal genome to study the evolution of small insertions and deletions in modern humans. BMC Evolutionary Biology 17: 179.
-
Das R, Sharma P (2020) Disorders of abnormal hemoglobin. In: Kumar D (Ed.), Clinical Molecular Medicine. Academic Press, pp: 327-339.
-
Feschotte C, Keswani U, Ranganathan N, Guibotsy ML, Levine D (2009) Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome biology and evolution 1: 205-220.
-
Gasparotto E, Burattin FV, Gioia VD, Panepuccia M, Ranzani V, et al. (2023) Transposable Elements Co- Option in Genome Evolution and Gene Regulation. Int J Mol Sci 24(3): 2610.
-
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, et al. (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66(3): 589-600.
-
Jilani M, Turcan A, Haspel N, Jagodzinski F (2022) Elucidating the Structural Impacts of Protein InDels. Biomolecules 12(10): 1435.
-
Jurka J, Bao W, Kojima KK, Kohany O, Yurka MG (2012) Distinct groups of repetitive families preserved in mammals correspond to different periods of regulatory innovations in vertebrates. Biology Direct 7(1): 36.
-
Kosmidis K (2025) Applied DNA visibility graphs: Understanding DNA structure-function relationship in genomics. Physica A: Statistical Mechanics and its Applications 663: 130436.
-
Kuhn GCS, Kuttler H, Moreira-Filho O, Heslop- Harrison JS (2012) The 1.688 Repetitive DNA of Drosophila: Concerted Evolution at Different Genomic Scales and Association with Genes. Molecular Biology and Evolution 29(1): 7-11.
-
Lillian P, Michael C, Jessica Q, Brian C, Isabel R, et al. (2021) An efficient method for simultaneous species, individual, and sex identification via in‐solution single nucleotide polymorphism capture from low‐quality scat samples. Molecular Ecology Resources 22: 1345-1361.
-
Lin M, Whitmire S, Chen J, Farrel A, Shi X (2017) Effects of short indels on protein structure and function in human genomes. Scientific reports 7(1): 9313.
-
Liu C, Tian Y, Zhang-xiong L, Yong-zhe G, Zhang B, et al. (2022) Identification and characterization of long- InDels through whole genome resequencing to facilitate fine-mapping of a QTL for plant height in soybean (Glycine max L. Merr.). Journal of Integrative Agriculture 21(7): 1903-1912.
-
Lugt RVD, Jacobs JJ (2026) Structural organization and function of telomeric chromatin. Nature Cell Biolog 28: 226-239
-
Magdalena T, Beltran A, Lehner B (2025) Deep indel mutagenesis reveals the impact of amino acid insertions and deletions on protein stability and function. Nature communications 16(1): 2617.
-
Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM (2018) Principles of adoptive T cell therapy in cancer. Semin Immunopathol 41: 49-58.
-
Miedl H, Dietrich B, Kaserer K, Schreiber M (2020) The 40bp Indel Polymorphism rs150550023 in the MDM2 Promoter is Associated with Intriguing Shifts in Gene Expression in the p53-MDM2 Regulatory Hub. Cancers (Basel) 12(11): 3363.
-
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, et al. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266(5182): 66-71.
-
Navarro LO, Varghese S, Han MV (2016) Measuring Accelerated Rates of Insertions and Deletions Independent of Rates of Nucleotide Substitution. J Mol Evol 83(3-4): 137-146.
-
Orkin SH, Kazazian HH (1984) The mutation and polymorphism of the human β-globin gene and its surrounding DNA. Annual Review of Genetics 18: 131- 171.
-
Pajnic IZ (2025) Analysis of Human Degraded DNA in Forensic Genetics. Genes 16(11): 1375.
-
Parasayan O, Laurelut C, Bôle C, Bonnabel L, Corona A (2024) Late Neolithic collective burial reveals admixture dynamics during the third millennium BCE and the shaping of the European genome. Sci Adv 10: eadl2468.
-
Parks MM, Lawrence CE, Raphael BJ (2015) Detecting non-allelic homologous recombination from high-throughput sequencing data. Genome Biology 16(72).
-
Rao SR, Trivedi S, Emmanuel D, Merita K, Hynniewta M (2010) DNA repetitive sequences-types, distribution and function: a review. J Cell Mol Biol 7(2): 1-11.
-
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, et al. (1989) Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 245(4922): 1066-1073.
-
Savino S, Desmet T, Franceus J (2022) Insertions and deletions in protein evolution and engineering. Biotechnology Advances 60: 108010.
-
Sehn JK (2015) Insertions and Deletions (Indels). In: Shashikant K (Eds.) Clinical Genomics. Academic Press, pp: 129-150.
-
Shapiro JA, von Sternberg R (2005) Why repetitive DNA is essential to genome function. Biological Reviews 80(2): 227-250.
-
Slean MM, Reddy K, Wu B, Edamura KN, Kekis M, et al. (2013) Interconverting Conformations of Slipped- DNA Junctions Formed by Trinucleotide Repeats Affect Repair Outcome. Biochemistry 52(5): 773-785.
-
Subramanian K, Chopra M, Kahali B (2024) Landscape of genomic structural variations in Indian population-based cohorts: Deeper insights into their prevalence and clinical relevance. HGG Adv 5(3): 100285.
-
Szymanska-Lejman M, Dziegielewski W, Dluzewska J, Kbiri N, Bieluszewska A, et al. (2023) The effect of DNA polymorphisms and natural variation on crossover hotspot activity in Arabidopsis hybrids. Nat Commun 14(33).
-
Uribe V, Badía-Careaga C, Casanova JC, Domínguez JN, de la Pompa, et al. (2014) Arid3b is essential for second heart field cell deployment and heart patterning. Development 141(21): 4168-4181.
-
Vecchio-Pagán B, Blackman SM, Lee M, Atalar M, Pellicore M J, et al. (2016) Deep resequencing of CFTR in 762 F508del homozygotes reveals clusters of non- coding variants associated with cystic fibrosis disease traits. Human genome variation 3(1): 1-9.
-
Wooster R, Bignell G, Swift S, Seal S, Mangion J, et al. (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378(6559): 789-792.
-
Zhang Y, Hulsman M, Salazar A, Tesi N, Knoop L, et al. (2025) Multi sample motif discovery and visualization for tandem repeats. Genome Res 35(4): 850-862.
-
Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, et al. (2014) Integrating human sequence datasets provides a resource of benchmark SNP and indel genotype calls. Nature Biotechnology 32(3): 246-251.
-
The Human Gene Mutation Database.
- Harnessing Molecular Glues for Next-Generation Vaccine, Cancer and Cardiovascular Disease Drug Development: A Comprehensive Review
- Lateral Cervical Epidermal Inclusion Cyst in a Paediatric Patient: A Rare Case Report
- Malarial Plasmodium Falciparum with Hepatitis B and C Virus Infections among Blood Donors in Ife Central Local Government Area, Ile Ife, Osun State, Nigeria
- Withanolides and Withaferin A- What’s next in Ashwagandha Research
- Designing of Dual Pulse Photoacoustic Tomography for Imaging of Drug-Response and Tumor Growth
- Trend Scenarios of Mortality due to Diabetes Mellitus and its Correlation with the Economic Sector, in the State of Mexico, for the Years 2020, 2025 and 2030