An Improved Protocol for the Expression and Purification of Tetracycline Monooxgenase: An Enzyme Involved in Antibiotic Resistance
Tetracyclines are an important group of antibiotics used in agricultural and clinical settings. They function by binding to the 30S subunit of the prokaryotic ribosome to effectively halt translation. The emergence of tetracycline resistant strains of pathogenic bacteria has threatened the continued use of the drug. One mechanism of tetracycline resistance is the degradation of the drug by the flavin dependent enzyme tetracycline monooxygenase (TetX). Only a limited number of studies of the enzyme have been conducted to date due to the limited amounts of pure protein that can be obtained from current protocols (~6 mg/L of cell culture). The current report describes the development of optimized expression and purification protocols to obtain larger amounts of pure, soluble enzyme suitable for detailed kinetic studies to deduce its chemical mechanism. During the initial phase of this work the pET22a+ plasmid with the gene encoding for TetX was used to transform either DH5α or Rosetta (DE3) pLysS Escherichia coli cells. Expression trials of these cell lines in which both the temperature and times of incubation with isopropyl β-D-1-thiogalactopyrandoside were then carried out. Of the conditions tested, optimal expression of TetX was found with a 20 h induction period at 30 °C using E. coli DH5α cells. Ammonium sulfate precipitation trials were then conducted where it was concluded that a 20 fold purification of the enzyme is achieved through treatment with 40% saturation of salt and collection of the supernatant after centrifugation. A nickel affinity chromatographic protocol was also developed, which purified the enzyme to high levels as judged by sodium dodecyl sulfate polyacrylamide gel electrophoresis and the specific activity of the purified sample. The expression and purification protocol developed here resulted in ~36 mg/L of cell culture, which is a 6-fold improvement over published protocols.
Introduction
Tetracyclines have been on the market for over 50 years and have been proven to be quite effective in treating many types of bacterial infections [1]. It is part of a group of important antibiotics that treat diseases ranging from acne to bacterial pneumonia. The efficacy of tetracycline has decreased in recent times [1]. This decrease is due to antibiotic resistance that many strains of bacteria have developed in response to the overuse of the drug and its release into the environment. In addition to active efflux and ribosomal protection mechanisms [1], many antibiotic resistant pathogens have evolved an enzymatic activity to degrade the drug in vivo [2, 3, 4]. Tetracyclines function by binding to the 30S subunit of the prokaryotic ribosome, which halts protein synthesis and prevents the growth of the bacterium [3]. An enzyme, known as tetracycline monooxygenase (TetX), chemically modifies the drug through the reaction shown in Scheme [1]. The enzyme is a flavin (FAD) and nicotinamide adenine dinucleotide phosphate (NADPH) dependent enzyme that is proposed to be a monooxygenase based on sequence and structural homology to other class A flavin monooxygeases [5]. The reaction produces 11a-hydroxytetracycline which is chemically unstable at physiological pH and decomposes to an unidentified mixture of products that are unable to bind to the ribosome to inhibit protein synthesis.3 Bacteria that have evolved this enzymatic activity have become resistant to tetracycline. This has dramatically decreased the effectiveness of the drug. Clearly, new strategies are needed to treat infections caused by tetracycline resistant pathogens.
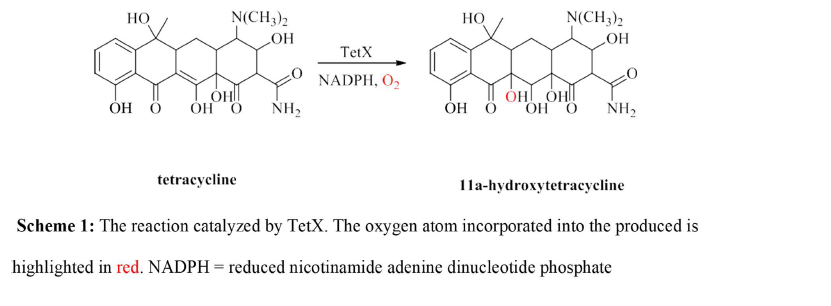
The inhibition of TetX through the design of mechanism- based inhibitors could provide one such strategy. This requires a detailed understanding of the catalytic mechanism of the enzyme which is currently unknown. To date only a limited number of kinetic studies needed to deduce the mechanism of TetX have been carried out due to the lack of a convenient expression and purification protocol to obtain large quantities of the enzyme. This account describes the development of such a protocol that involves the expression of the recombinant enzyme through the induction of Escherichia coli DH5α, an ammonium sulfate precipitation and nickel affinity column chromatography. The protocol resulted in 36 mg of TetX from 1 L of culture, which is a 6-fold improvement from the previously reported procedure.3 The procedure described herein will allow for the production of large amounts if pure TetX for mechanistic studies that could lead to the development of new drugs to be used in conjugation with tetracyclines to treat bacterial infections.
Materials and Methods
Materials
The pET22a+ plasmid containing the gene encoding for TetX was a generous gift from Professor Gerard Wright of McMaster University, Ontario, Canada. DH5α and Rosetta (DE3) pLysS Escherichia coli competent cells were provided by Professor Giovanni Gadda of Georgia State University, Atlanta, GA. All other reagents were of the highest purity commercially available.
Induction Trials
The pET22a+ plasmid containing the gene encoding TetX was used to transform either DH5α or Rosetta (DE3) pLysS competent cells using the heat shock method of Inoue, et al. [6]. The cells were plated onto Luari-Bertani (LB) agar containing 34 g/mL kanamycin and 50 g/mL ampicillin and were incubated overnight at 37°C. Single colonies were then used to inoculate 50 mL LB supplemented with 50 g/mL kanamycin and the cells were grown to an optical density at 600 nm (OD600) of between 0.6-0.8. When the appropriate OD600 was reached the cells were induced through the addition of a final concentration of 1 mM isopropyl β-D-1-thiogalactopyrandodice (IPTG). To optimize the amounts of soluble and active TetX obtained from pET22a+-TetX transformed competent cells the following induction conditions were tested: 5 h incubation at 30°C; 20 h incubation at 30°C; 5 h incubation at 37°C and 20 h incubation at 37°C. Parallel experiments using either DH5α or Rosetta (DE3) pLysS were carried out to determine which cell strain was better to produce TetX. In each experiment, cells were harvested, lysed and used to determine the specific TetX activity as described below.
Cell Lysis and Specific Activity Determination
Cells were harvested through centrifugation at 12,000 x g at 4°C for 20 min and were resuspended in 50 mM sodium phosphate, 100 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid and 10% glycerol pH 7.0.
Lysis was achieved through incubation with 1 mg/mL lysozyme on ice for 30 min on a stir plate. Addition of FAD to a final concentration of 100 µM was found to significantly improve TetX activity of the cell extracts so the cofactor was included in both the induction trials and the purification of the enzyme. After incubation with lysozyme, the extract was centrifuged at 4°C and 12,000 x g for 20 min. The supernatant was then analyzed for TetX activity and total mass of protein. TetX activity was measured using a Clarke type oxygen electrode (Hansatech Instruments, Norfolk, United Kingdom). Assays were conducted in 50 mM sodium phosphate pH 6.0 using 1 mM tetracycline hydrochloride and 100 µM NADPH at room temperature. Consistent with the observations of Yang, et al. [3] no activity was observed with NADH. Assays were conducted in triplicate and the average value is reported with its standard deviation. Tetracycline was prepared in 100% ethanol. The total concentration of ethanol in the assay mixture was maintained at 1% to minimize its effect on enzyme activity. The electrode reading was corrected for the volume of extract added to the reaction mixture to calculate the total units of TetX activity (U total). The total mass of protein in the cell extracts was determined using the method of Bradford [7] with bovine serum albumin as standard. The activities of the extracts were then divided by the total mg of protein to calculate a specific TetX activity (U/mg).
After optimal expression conditions were found, a colony screen of DH5α-TetX was carried out by plating cells onto LB agar containing 34 g/mL kanamycin and 50 g/mL ampicillin. The cells were incubated overnight at 37°C after which approximately 75 single colonies formed. Ten colonies were picked at random and used to express TetX using the optimal conditions identified above. Replica plates were made for each of the 10 cultures and the specific TetX activities were determined as described in Cell Lysis and Specific Activity Determination. The colony that was found to have the highest U/mg was used to prepare permanent glycerol stocks that were stored as 1 mL aliquots at -80°C until use. All subsequent experiments were carried out through a 20 h induction of these stocks at 30°C with 1 mM IPTG.
Ammonium Sulfate Precipitation
A 1.2 L culture of LB containing 34 g/mL kanamycin and 50 g/mL ampicillin was inoculated with DH5α-TetX and cell extracts were prepared as described above. The extract was divided into 6 x 190 mL fractions that were treated with 0, 20, 40, 60, 80 or 100% saturation with ammonium sulfate. The sample was incubated with ammonium sulfate for 30 min on ice then centrifuged at 12,000 x g for 20 min at 4°C. The supernatant was separated from the pellet. Each pellet was subsequently re suspended in 50 mM sodium phosphate pH 7.0. All samples were then dialyzed against 50 mM sodium phosphate pH 7.0 + 10% glycerol for four changes of at least 1 h. The specific activities were then determined for each fraction by determining the TetX activity as described above and quantifying the total mass of protein. Significant loss of the flavin cofactor was observed, but activity was restored upon addition of a final concentration of 1 mM FAD.
Nickel Affinity Column Chromatography
A 1 L culture of DH5α-TetX was induced, lysed and treated with 40% saturation of ammonium sulfate as described above. After dialysis against 50 mM sodium phosphate + 10% glycerol pH 7.4, the sample was mixed with an equal volume of 50 mM sodium phosphate + 200 mM sodium chloride + 10% glycerol pH 7.4 and loaded onto a 10 mL nickel sepharose column equilibrated with the same buffer. The column was washed with 2 bed volumes of buffer before eluting with 4 bed volumes of 250 mM imidazole. Fractions were collected in 5 mL increments and analyzed by the Bradford assay and activity with 1 mM tetracycline + 100 µM NADPH at pH 6.0 and room temperature. Fractions containing TetX were pooled, dialyzed against 50 mM sodium phosphate + 10% glycerol and stored at -80°C until use. Protein purity was judged through sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using a RunBlue™ TEO-Tricine gel (Expedeopn, San Diego, CA) following the manufacturer’s instructions.
Results and Discussion
Induction Trials and Preparation of Permanent Glycerol Stocks The overall goal of this work was to develop a protocol that results in larger amounts of TetX than that described previously (~6 mg/L of cell culture) [3]. To achieve this goal, a series of experiments were conducted in which the cell strain, induction time and temperature were varied to determine optimal conditions for the expression of TetX. Under all conditions tested the specific activity (U/mg) was higher with DH5α competent cells as opposed to Rosetta (DE3) pLysS (Table 1). Specific activity was maximal when the cells were induced with 1mM IPTG and incubated for 20 h at 30°C (730 ±10 U/mg). While both and costs limited the trials to only eight conditions, induction at 30°C for 20 h produced a ~7x higher specific activity than the least optimal conditions.
| Condition | Units of Activitya U | Total Proteinb mg | Specific Activity U/mg |
|---|---|---|---|
| Rosetta (DE3) pLysS | |||
| 37 ºC for 20 h | 350 ± 60 | 1.12 | 310 ± 50 |
| 37 ºC for 5 h | 275 ± 40 | 2.16 | 130 ± 20 |
| 30 ºC for 20 h | 310 ± 30 | 0.999 | 310 ± 30 |
| 30 ºC for 5 h | 85 ± 10 | 0.828 | 100 ± 10 |
| DH5α | |||
| 37 ºC for 20 h | 480 ± 10 | 1.33 | 360 ± 10 |
| 37 ºC for 5 h | 465 ± 30 | 1.99 | 230 ± 10 |
| 30 ºC for 20 h | 650 ± 10 | 0.885 | 730 ± 10 |
| 30 ºC for 5 h | 475 ± 25 | 4.4 | 110 ± 10 |
Table 1: Induction Trials for the Expression of TetX. a: TetX activity was measured in 50 mM sodium phosphate pH 6.0 at room temp
Table 1: Induction Trials for the Expression of TetX. a: TetX activity was measured in 50 mM sodium phosphate pH 6.0 at room temperature using 1mM tetracycline and 100 µM NADPH. Assays were conducted in triplicate and the average value is reported with its standard deviation. b: Total protein was quantified using the Bradford assay with bovine serum albumin as standard.
Once optimal expression conditions were identified, a colony screen was conducted in which DH5α was plated onto LB agar containing 34 g/mL kanamycin and 50 g/mL ampicillin? Single colonies were used to inoculate LB broth containing the same antibiotics. Cells were induced with 1 mM IPTG for 20 h at 30ºC, harvested and lysed to prepare cell extracts. The results are summarized in Table 2. The specific activities ranged from ~50-3210 U/mg. This variability is common and emphasizes the importance of conducting a colony screen prior to embarking on the development of a large-scale purification protocol. The colony screen was repeated with two independent groups of students. While only one set of experiments are shown for clarity, the results obtained with each group of students were qualitatively the same demonstrated a reproducibility of the results obtained.
| Colony | Units of activity, U | Total protein mg | Specific activity U/mg |
|---|---|---|---|
| 1 | 40 ± 2 | 0.174 | 230 ± 10 |
| 2 | 30 ± 3 | 0.656 | 45 ± 5 |
| 3 | 30 ± 8 | 0.381 | 80 ± 20 |
| 4 | 220 ± 10 | 0.135 | 1630 ± 75 |
| 5 | 40 ± 6 | 0.546 | 75 ± 10 |
| 6 | 40 ± 12 | 0.516 | 80 ± 25 |
| 7 | 575 ± 11 | 0.179 | 3210 ± 55 |
| 9 | 95 ± 9 | 0.437 | 80 ± 10 |
| 10 | 510 ± 40 | 0.521 | 100 ± 10 |
Table 2: Colony Screen for TetX Activitya.
Development of a Polagraphic TetX Activity Assay
Previous studies of TetX utilized activity assays that followed the absorbance changes of the NADPH cofactor that occurs upon its oxidation during catalytic turnover.3 Such experiments are cumbersome and too time consuming for detailed studies of the kinetic mechanism of the enzyme, which are planned to initiate the development of new drugs. A polarographic assay was therefore developed using a Clark type oxygen electrode. This assay is commonly used in the study of flavin dependent enzymes [8, 9]. While the assay is routinely used in other systems, considerable effort was aAll colonies were DH5α-TetX induced at 30ºC for 20 h. 100 mM FAD was included in the lysis buffer and the activity was measured in triplicate and the average ± standard deviations are reported needed in order to obtain reliable and reproducible traces to test the expression systems used in this study. Initial attempts to detect TetX activity resulted in limited oxygen consumption and traces such as that shown in Figure 1a were obtained. DNA sequencing of plasmids extracted from the cell constructs confirmed the presence of the tetX2 gene and the activity observed was clearly not an artifact as evident from the complete lack of oxygen consumption observed in extracts of cells lacking the tetX2 gene (data not shown). The initial rates of oxygen consumption were ~2 s-1 and went to 0 after ~ 1 minute. These low rates of activity and pale-yellow color of the extracts suggested a low flavin to protein stoichiometry. To test for this possibility, a culture of DH5 -TetX was induced with IPTG under the optimized conditions described above and split into equal volumes. One of the samples was lysed in the absence of exogenous FAD while the other was incubated with a final concentration of 1 mM FAD. As shown in Figure 1b, the cell extract obtained upon incubation with the FAD cofactor was significantly more active (~15-fold higher U/mg) than that obtained without inclusion of FAD in the lysis buffer? Even more importantly, the trace in Figure 1 gave an error that was less than 10% that of the baseline. This will allow for the accurate determination of both the kcat and kcat/Km values of the enzyme with either tetracycline oxygen or NADPH as substrate. It also allowed for reliable results to be obtained here for both the expression and purification of the enzyme.
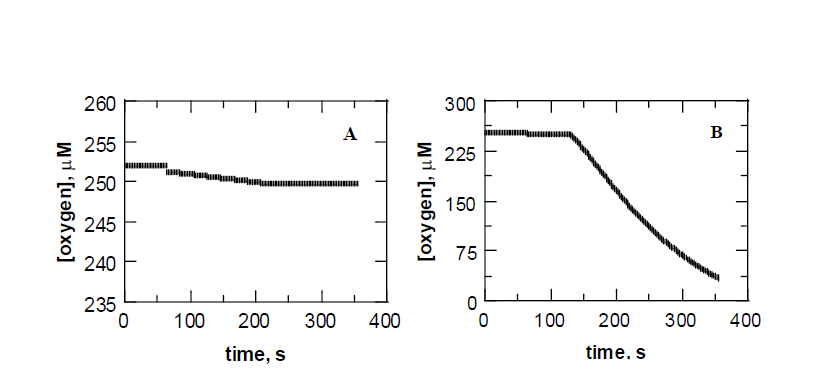
Figure 1: Activity assays of TetX in 50 mM sodium phosphate pH 6.0 and room temperature. The reaction contained 1 mM tetracycline and 100 M NADPH and was initiated by the addition of the cell extract. Assays were typically repeated in triplicate and the average values are reported in Tables 2 and 3. Panel A shows a typical trace collected without inclusion of FAD in the lysis buffer. Panel B show a typical trace obtained after inclusion of 1 mM FAD in the lysis buffer.
While exhaustive studies to determine the source of the dramatic increase in oxygen consumption observed upon exogenous addition of FAD were not within the scope of the goals of this work, a reasonable hypothesis to explain the results can be postulated based on X-ray crystallographic structures of the enzyme [2, 10, 11] and the observations made while dialyzing samples during the ammonium sulfate and nickel affinity column chromatography steps (see below). The FAD cofactor is non-covalently bound to the enzyme and the crystal structure of the protein in complex with 7-chlorotetracyline (a substrate analog) shows that FAD sits in between two domains in each subunit of the dimer. While the Roseman fold that is typical of flavoenzymes is observed to interact the nucleotide moiety of the cofactor, the isoalloxine ring is exposed to an opening between the domains. In the absence of substrate, partial unfolding may result in a release of the cofactor, which would be exacerbated by the vigorous resuspension of the cell pellets needed during the lysis procedure. Consistent with these observations the buffer solution turned bright yellow after the first round of dialysis as would be expected if FAD is released from enzyme since its mass is well below the cutoff of the membranes used in this study. Inclusion of exogenous FAD would shift the equilibrium of the system to holoenzyme and would also provide numerous electrostatic and hydrogen bonding intractions with the protein to stabilize its overall structure. It is important to note that, as expected, no oxygen consumption was observed when free FAD was mixed with tetracycline under the same reaction conditions.
Regardless of the exact reason for the observations of Figure 1, it is clear that exogenous FAD should be included during the expression and purification of the enzyme to stabilize its activity. This was done in all subsequent experiments carried out in this study.
| Specific Activity of Supernatant (U/mg) | Specific Activity of Pellet (U/mg) | |
|---|---|---|
| 0 | (2.8 ± 0.10) x 103 | ---b |
| 20 | (2.6 ± 0.61) x 103 | 0b |
| 40 | (5.7 ± 0.61) x 104 | 0c |
| 60 | (2.0 ± 0.10) x 103 | (7.0 ± 0.2) x 102 |
| 80 | (4.4 ± 0.28) x 102 | (2.1 ± 0.3) x 103 |
| 100 | (2.1 ± 0.20) x 101 | (2.7 ± 0.11) x 103 |
Table 3: Ammonium Sulfate Fractionationa. a: DH5α-TetX was induced at 30ºC for 20 hr. 100 µM FAD was included in the lysis buffer
Table 3: Ammonium Sulfate Fractionationa. a: DH5α-TetX was induced at 30ºC for 20 hr. 100 µM FAD was included in the lysis buffer, which was split into 6 equal volumes and treated with increasing percent saturation of ammonium sulfate. Activity was measured in triplicate and the average ± standard deviations are reported. b: No pellet formed. c: No activity was detected in the re suspended pellet.
Ammonium Sulfate Precipitation
A 1.2 L sample of LB containing 34µg/mL kanamycin and 50 µg/mL ampicillin was inoculated with DH5α and the lysate was split into separate fractions and treated with increasing amounts of ammonium sulfate. Table 3 shows the total units measured using the polagraphic activity assay, the total mass of protein determined from the Bradford assay and the calculated specific activity at each percent saturation of ammonium sulfate. The results showing no activity in the protein pellet below 40% saturation and a fractionation of TetX activity between the supernant and pellet at percent saturations above 40% suggested that value is an appropriate cut to partially purify the protein before column chromatography. Including this step before loading extracts onto the column reduces the total amount containment proteins and thereby the amount of nickel NTA resin that is needed, which significantly saves both time and money.
| Total Activity (μmolO2/s)a | Total protein (mg)b | Specific Activity (U/mg)c | Purification Index | |
|---|---|---|---|---|
| Cell Extract | (6.1 ± 0.1) x 10541,000 | 165 ± 5 | (3.7 ± 0.3) x 103 | 1 |
| 40% (NH4)2SO4 | (7.8 ± 0.1) x 106 | 105 ± 2 | (7.4 ± 0.1) x 104 | 20 |
| Nickel Affinity | (1.0 ± 0.1) x 107 | 36 ± 1 | (2.8 ± 0.1) x 105 | 75 |
Table 4: Purification of Recombinant TetX. a: Assay contained 1 mM tetracycline + 50 µM NADPH in 50 mM sodium phosphate at pH 6.0
Table 4: Purification of Recombinant TetX. a: Assay contained 1 mM tetracycline + 50 µM NADPH in 50 mM sodium phosphate at pH 6.0. Assays were conducted in triplicate wand the average value is reported with its standard deviation. b: Mass of protein was determined by Bradford assay using bovine serum albumin as a standard protein. The assay was conducted in duplicate and the average value is reported with its standard deviation. c: Specific activity was determined by the division of units of activity and total protein Nickel Affinity Chromatography and Large Scale TetX Protein Purification. A 1 L culture of DH5α-TetX was induced, lysed and treated with 40% saturation of ammonium sulfate as described above. The pellet was discarded and the supernant was dialyzed and loaded onto a nickel affinity column for purification as described in the methods section. Fractions that contained TetX as judged by the activity assay were pooled, dialyzed against 50 mM sodium phosphate +10% glycerol pH 7.0 and stored at -80 oC. A Bradford assay revealed that a total mass of 36 mg of pure TetX was obtained by the procedure described here, which is a 6-fold improvement over the previously described procedure. Comparison of the specific activity of the purified sample to that of the cell extract demonstrated that both the ammonium sulfate fraction and the affinity purified the sample 75 times that of the cell extract (as determined through a comparison of specific activities). SDS-PAGE analysis showed a single band after column chromatography which substantiates the large purification index calculated. The data from the purification of the enzyme from DH5α-TetX is shown in Table 4.
Conclusion
The emergence of tetracycline resistant bacteria has been an increasing concern in both medicine and agriculture [1] which necessitates new treatment strategies to fight disease. A prevalent mechanism of antibiotic resistance is the degradation of the drug by TetX [3, 4]. The current work describes an optimized expression and purification protocol that resulted in a significantly larger amount of pure enzyme that is suitable for detailed mechanistic studies than that obtained previously [3]. Such studies will utilize non-competitive kinetic isotope effects [9, 12, 13] with tetracycline and NADPH both in steady state and pre-steady state to obtain information regarding the transition state structure of the TetX reaction. The protocol described here can easily be extended to any site-specific mutants that may be created based on the wealth of structural information known for TetX [2, 10, 11] to further probe the transition state of the reaction. Once a detailed picture emerges, the tools of synthetic organic chemistry can be applied to generate transition state analogues that are expected to be potent inhibitors of the enzyme.
Such a drug design strategy has been successfully applied to inhibit critical metabolic pathways in MpnE [14] and purine-nucleotide phosphorylase [15]. Inhibitors created from mechanistic insights of TetX would then be used in conjugation with existing tetracyclines that are currently on the market to treat infectious disease. While several important studies were previously reported for TetX [2, 10, 11] an extensive mechanistic characterizations of the enzyme have been limited by the low yields obtained by previously reported expression and purification protocols. The current account will break down these barriers, allow for detailed kinetic studies an should usher a new avenue of TetX research.
Acknowledgment
This work was supported by the University Research Award from Texas A&M University-Kingsville Texas Compressive Research Fund, a Departmental Research Grant from The Robert A. Welch Foundation AC-0006 and the Ronald E. McNair Program. Profs. Gerard Wright (McMater University, Ontario Canada) and Giovanni Gadda (Georgia State University, Atlanta GA) are gratefully acknowledged for supplying the pET 22a+ plasmid containing the TetX gene and E. coli competent cells, respectively.
References
-
Speer BS, Shoemaker NB, Salyers AA (1992) Bacterial resistance to tetracycline: mechanisms, transfer, and clinical significance. Clin Microbiol Rev 5(4): 387-399.
-
Volkers G, João M Damas, Gottfried J Palm, Santosh Panjikar, Cláudio M Soares, et al. (2013) Putative dioxygen-binding sites and recognition of tigecycline and minocycline in the tetracycline-degrading monooxygenase TetX. Acta Crystallogr D Biol Crystallogr 69(9): 1758-1767.
-
Yang W, Ian F Moore, Kalinka P Koteva, David C Bareich, Donald W Hughes, et al. (2004) TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem 279(50): 52346-52352.
-
Ian F Moore, Donald W Hughes, Gerard D Wright (2005) Tigecycline is modified by the flavin- dependent monooxygenase TetX. Biochemistry 44(35): 11829- 11835.
-
Mieke ME Huijbers, Stefania Montersino, Adrie H Westphal, Dirk Tischler, et al. (2014) Flavin dependent monooxygenases. Arch Biochem Biophys 544: 2-17.
-
Inoue H, Nojima H, Okayama H (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96(1): 23-28.
-
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
-
Chapman SK, Reid GA (1999) Flavoprotein Protocols. In: Walker JM, Methods in Molecuar Biology, Humana Press.
-
Francis K, Gadda G (2019) On the use of noncompetitive kinetic isotope effects to investigate flavoenzyme mechanism. Methods Enzymol 620: 115-143.
-
Gesa Volkers, Gottfried J Palm, Manfred S Weiss, Gerard D Wright, Winfried Hinrichs (2011) Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett 585(7): 1061-1066.
-
Gesa Volkers, Linda Schuldt, Gottfried J Palm, Gerard D Wright, Winfried Hinrichs (2010) Crystallization and preliminary X-ray crystallographic analysis of the tetracycline-degrading monooxygenase TetX2 from Bacteroides thetaiotaomicron. Acta Crystallogr Sect F Struct Biol Cryst Commun 66(Pt 5): 611-6144.
-
Cleland WW (1982) The use of isotope effects to determine transition-state structure for enzymic reactions. Methods Enzymol 87: 625-641.
-
Kohen A, Limbach HH (2005) Isotope Effects in Chemistry and Biology, CRC Press, Boca Raton, FL, pp; 1096.
-
Joshi S, Dmytro Fedoseyenko, Nilkamal Mahanta, Rodrigo G Ducati, Mu Feng, et al. (2019) Antibacterial Strategy against H. pylori: Inhibition of the Radical SAM Enzyme MqnE in Menaquinone Biosynthesis. ACS Med Chem Lett 10(3): 363-366.
-
Evans GB, Schramm VL, Tyler PC (2015) The Immucillins: Design, Synthesis and Application of Transition- State Analogues. Curr Med Chem 22(34): 3897-3909.
- Origin, Evolution, and Functional Impact of Short Insertion- Deletion Variants in Human Genomes: A Review
- Harnessing Molecular Glues for Next-Generation Vaccine, Cancer and Cardiovascular Disease Drug Development: A Comprehensive Review
- Lateral Cervical Epidermal Inclusion Cyst in a Paediatric Patient: A Rare Case Report
- Malarial Plasmodium Falciparum with Hepatitis B and C Virus Infections among Blood Donors in Ife Central Local Government Area, Ile Ife, Osun State, Nigeria
- Withanolides and Withaferin A- What’s next in Ashwagandha Research
- Designing of Dual Pulse Photoacoustic Tomography for Imaging of Drug-Response and Tumor Growth