A Battle Between Pathogens and Medicinal Chemistry: Tetracycline Generations and the Evolution of Resistance Mechanisms
Tetracyclines have been utilized extensively for more than 70 years which has led to the development of resistance by pathogenic bacteria. New generations of tetracyclines have been produced in response to this resistance. Since tetracyclines are broad-spectrum antibiotic used to treat different bacterial infections, it is essential for clinical use. Synthetic strategies have been developed to modify the drug in response to resistance. Currently, three different third-generation tetracyclines are on the market and are sold as Tygacil, Nuxga and Xerara. These tetracyclines are much more effective than previous generations due to an increased affinity towards the target ribosome and an ability to evade bacterial efflux proteins. However, at least for the case of Tygacil, which has been tested in vitro, these tetracyclines might be susceptible to degradation by the enzyme tetracycline monooxygenase (TetX), which modifies the antibiotics to 11a-hydroxytetracycline. The TetX reaction product is unstable at physiological pH and decomposes before it can interact with the bacterial ribosome to inhibit protein synthesis. Understanding details about the catalytic mechanism of TetX could lead to the development of inhibitors of the enzyme to provide an additional strategy to combat antibiotic resistance. The current review will detail the chemical features of each generation of tetracyclines, the biochemical basis for tetracycline treatment and the mechanisms by which bacteria are able to resist the drugs. It will end with a discussion of TetX and an activity assay developed to study the enzyme.
Introduction
Tetracycline antibiotics have been utilized since 1948 [1, 2]. It all started with a young boy, Toby Hockett, being treated with chlortetracycline (trade name Aureomycin) for a serious infection after surgery [2]. During the mid to late 1940s, not many antibiotics were proven to be useful in treatment of bacterial infections. Aureomycin was still under evaluation for medicinal use in humans, but the parents of Toby decided to move forward with treatment since there were no other viable options. This antibiotic helped him to fully recover from the infection within months of treatment, which thus began the clinical use of tetracycline antibiotics. Tetracyclines have been used to treat different gram-positive and gram-negative bacteria, and pathogens like protozoa [1]. It is used extensively for treatment of urogenital tract infections, periodontal and Lyme disease, rickettsia, and bacterial respiratory tract infections. Tetracycline antibiotics are not only used for human pathogens, but have also been in agriculture to prevent bacterial contamination and in the production of animal food industry products [1]. From the use of tetracycline antibiotics for over 70 years, resistance towards this drug has developed. This review will describe the biochemical basis of tetracycline treatment, the mechanisms of antibiotic resistance evolved by pathogens and the development of new generations of drugs to overcome this resistance.
The name tetracycline derives from the structure of its four six-membered ring naphthalene core (Figure 1) [5]. The minimal pharmacophore of tetracycline antibiotics showing medicinal use contains 6- deoxy-6-demethyltetracycine (i.e. all R groups in Figure 1 are H atoms) [3]. Naturally occurring tetracyclines are highly oxidized and only show variability on three of the carbon atoms in the ring system (C5-C7 in Figure 1). They were first isolated from Streptomyces sp. in the 1940s and represent the first generation of the drug. Examples include chlortetracycline (first reported in 1948), oxytetracycline (first reported in 1950) and tetracycline (first reported in 1953) [1]. Figure 1 illustrates the chemical structure of tetracycline antibiotics along with the accepted numbering system and letter designation of the four rings of the naphthalene core of the antibiotics. This system will be used in subsequent sections of the review. Semi- synesthetic tetracyclines have been developed over the past several decades through modifications of the substituents highlighted in the boxed regions of Figure 1. These were done in response to tetracycline resistance as described below.
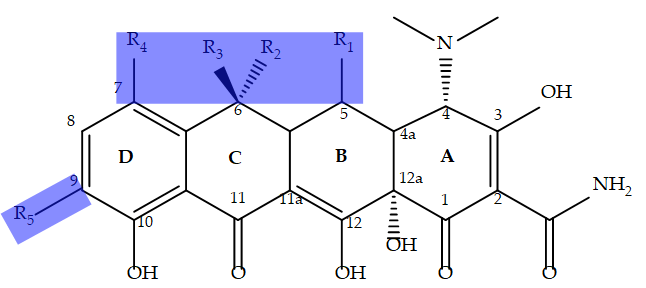
Figure 1: Tetracycline Pharmacophore. The basic structure of tetracycline antibiotics consists of four six membered rings. The substituents outside the boxed region are invariant in all medicinally active tetracyclines. The boxed regions show substituents that have been modified to produce new generations of tetracyclines in response to the emergence of resistant pathogens as discussed below. The numbering and letter designations of the rings are based what is described in the literature [3, 4, 5].
Tetracycline antibiotics bind to the 30S subunit of the prokaryotic ribosome to prevent the association of amino-acyl tRNA during translation [1, 6, 7]. Thus, the drugs effectively prevent protein synthesis and exert a bacteriostatic effect. The prevention of cellular growth by the action of tetracycline antibiotics allows a patient’s innate immune system to clear the bacteria and recover from an infection. The drug has been quite effective in treating a variety of diseases and is given prophylactically to those who are immunocompromised. They are also given as additives to animal feeds in agriculture to serve as growth promoters for production use. These production uses are not intended to manage a specific disease that may be ongoing or at risk of occurring, but rather are expressly indicated and used for the purpose of enhancing the production of animal- derived products (e.g., increasing rate of weight gain or improving feed efficiency). It is essential to pursue prudent use of tetracyclines to minimize resistance and ensure their continued effectiveness and availability of these products and to curtail impacts on human health [8]. Despite this prudence, the widespread use of tetracycline antibiotics for over 70 years has inevitably led to resistance towards the drug by pathogens.
Resistance has developed by different mechanisms [1, 5, 9, 10]. These include ribosomal protection, efflux and enzymatic degradation of the drug. Ribosomal protection involves proteins that block tetracyclines from binding to the ribosome. Efflux proteins also physically transport the antibiotic outside the cell thereby preventing it from halting translation by the pathogen. A less prevalent, but increasingly concerning mechanism of tetracycline resistance is enzymatic inactivation through chemical modification of the antibiotic [3]. One such enzyme is tetracycline monooxygenase (TetX) which uses flavin adenine dinucleotide (FAD) and reduced nicotinamide adenine dinucleotide phosphate (NADPH) to degrade tetracycline [11, 12]. This completely prevents the inhibition of protein synthesis in pathogenic bacteria [12]. The enzyme was first found in an obligate anaerobe, Bacteroides fragilis, and cloning of the gene encoding for the enzyme into Escherichia coli was shown to provide the bacterium resistance to tetracycline [3, 11, 12, 13]. It is feared that gene transfer from soil microbes to pathogens will lead to more widespread resistance to tetracyclines which will decrease the effectiveness of this class of antibiotics.
This review starts with a description of the chemical structures of the multiple generations of tetracyclines developed to date in response to antibiotic resistance in pathogens. Details on the mechanism by which tetracycline antibiotics treat disease will then be discussed followed by the strategies pathogens have evolved to resist the drug. Special emphasis will be placed on the enzyme TetX and the results recently obtained to allow for a detailed characterization of the enzyme in order to understand its chemistry. The review ends with concluding remarks and a summary of the ways in which synthetic strategies have improved the efficacy of the drug. It is hoped this review will raise awareness to the increased problems of tetracycline resistance in human and agricultural pathogens.
Generations of Tetracycline
As illustrated in Figure 2, all clinically used tetracyclines are characterized by different functional groups among the four rings of the naphthalene core [1, 5, 10]. To simplify the presentation only a select number of tetracyclines are presented as representative examples of each generation and the regions that were most commonly modified are highlighted in the boxed regions. As seen in Figure 2, the most effective modifications made to overcome resistance occurred on rings B-D at carbons 5, 6 and 7. Modifications were also made in other regions, but these gave rise to decreased antimicrobial activity as compared to natural tetracyclines (A in Figure 2) [2, 5], whereas the functional groups modified in the upper periphery was shown to increase antimicrobial activity [9, 12]. The emergence of clinical isolates showing tetracycline resistance in the 1950s and 1960s prompted medicinal chemistry in both industry and academia to develop semi- synesthetic approaches to battle resistance and treat infection. Second generation tetracyclines focused on regions of the molecule that are varied in naturally occur tetracyclines found in Streptomyces, namely the substituents on C5, C6 and C7. These tetracyclines remained effective in treating disease for around 20 years, but bacterial pathogens fought back by adapting to these drugs as well. This was achieved through the development of resistance through the same mechanisms as with the first generation of the drug and led to the development of more modern tetracyclines. Modern versions started with the basic scaffold of minocycline, but contain additional modifications at C9.
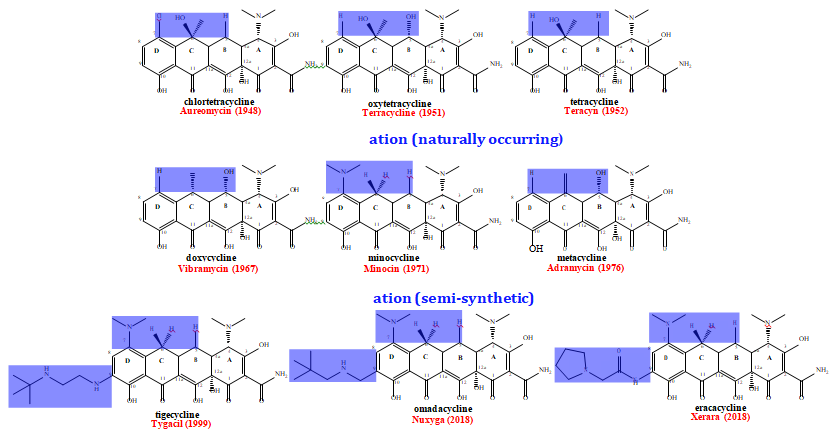
3rd Generation (glycylcyclines). Figure 2: Representative Tetracyclines from Different Generations. A) The first generation of the drug were isolated directly from different strains of the Streptomyces genus and were put into clinical use during the 1940’s through the 1950’s. Development of clinical resistance led to semi-synesthetic routes to generation subsequent generations. B) Second generation tetracyclines were produced through the modification of substituents on C5-7. These came onto the market in the late 1960s through the 1970s. C) More modern generations were produced through the addition of substitutes to C9 and came onto the market in the late 1990s through the 2010s.
The first compound belonging to the tetracycline family was chlortetracycline and was discovered by Dr. Benjamin Duggar. Chlortetracycline contains a chlorine atom at C7 of ring D of the naphthalene core (Figure 2) and has the trade name Aureomycin. The discovery and demonstrated clinical effectiveness of chlortetracycline led more companies to search for antibiotics in soil microbes. A ground sample isolated by Pfizer discovered a compound that was very similar to chlortetracycline [2]. After combining resources, the efforts of the Pfizer team and Robert B. Woodward of Harvard University, were successful in outlining the similarities and differences between chlortetracycline and another naturally occurring tetracycline known as oxytetracycline (Figure 2). Oxytetracycline (trade name Terramycin) contains a hydroxyl group attached to C5 (ring B in Figure 1) [2]. “Tetracycline” was created after modification of oxytetracycline [4]. Tetracycline is the result of dehydroxylating C5 of the naphthalene ring to mimic what is found in chlortetracycline as shown in Figure 2 [2]. These three tetracyclines comprise the first generation of the drug and were quite effective in treating bacterial infections for around 30 years until resistance diminished the efficacy of the drug.
Medicinal chemists continued to take semi-synthetic approaches to tackle the problem of bacterial resistance towards tetracyclines. This occurred through the generation of a second generation of the drug starting in the late 1960s and continuing through the 1970s. These tetracyclines include metacycline (trade name Adramycin), doxycycline (trade name Vibramycin), demecycline (trade name Declomycin), sanocycline (trade name Bonomycin) and minocycline (trade name (Minocin). Pfizer chemists modified the ring C of the naphthalene core of tetracycline with the goal of keeping chemical stability, but overcoming bacterial resistance [2]. Doxycycline was then produced by Charlie Stephens and was approved by the food and drug administration soon after for clinical use. After isolating more soil samples, demeclocycline was discovered, which lead to sancycline production through modifications to the C and D rings of demeclocycline. One of the most effective second-generation tetracycline is minocycline that was produced through the modification of the D ring of sancycline at C7 Figure 2). As with the first generation, second generation tetracyclines proved effective for around 30 years until pathogenic bacteria became highly resistant to the drugs making them less useful in clinical settings.
This led to more modern generations of tetracyclines that were developed by focusing more on the C9 position of ring D in the naphthalene core [4]. Tetracyclines of this generation retained the diethylamino moiety at C7 that is found in minocycline since the substituent proved effective in ribosomal binding to bacteria. Representative third generation tetracyclines are illustrated in Figure 2. Since many of these third-generation tetracyclines were synthesized with a glycyl moiety at C9 they are often called glycylcyclines [4, 5]. These modern tetracyclines are more effective in the treatment of both gram-positive and gram-negative bacteria. The first third generation tetracycline was tigecycline which was developed with the hopes of stopping resistance from occurring so quickly. This drug appeared on the market around the turn of the century, remains in clinical use and is sold with the trade name Tygacil [5]. The most recent glycylcyclines to come onto the market are omadacyclcine and eravacycline that both became available in 2018 and are sold as Nuxyga and Xerara, respectively. While these third- generation antibiotics do not appear to be as susceptible to the resistance mechanisms seen in earlier generations the battle does not seem to be over just yet. At least in the case of tigercycline, which is the only third generation tetracycline for which biochemical data is currently available, the drug is subject to enzymatic degradation by flavin dependent monooxygenases [3, 11, 12]. This coupled with the observations that bacterial pathogens were able to readily adapt to previous generations of tetracyclines within 20-30 years demonstrates that biomedical experiments is needed to effectively treat infection.
Tetracycline: Mode of Action
Tetracycline antibiotics prevent translation from occurring in prokaryotes thereby preventing bacterial cell growth [1, 2]. Protein synthesis is inhibited through the binding of the naphthalene core of the antibiotic to prokaryotic ribosomal proteins. Binding experiments reveal that the 30S ribosomal subunit has a higher affinity for the drug than either the 50S subunit or the entire intact ribosome [1, 14]. Ambiguity seems to exist on the nature of tetracycline binding to the 30S ribosomal subunit. Based on the observation of up to five distinct tetracycline binding sites in the solved structures of the Thermus thermophilus ribosome cooperative binding of tetracyclines was suggested [1, 15, 16]. However, the electron density in many of the proposed binding sites was poor. More importantly, direct measurements of equilibrium binding constants through fluorescence titrations of a number of tetracyclines with the ribosome revealed saturation curves with Hill coefficients very close to 1, strongly implicating non-cooperative binding [14]. Subsequent molecular dynamics simulations further supported non-cooperative binding of tetracycline to a single high affinity binding site (the so called “primary binding site”) [17]. Tetracycline is able to effectively bind to this site with KD values in the 10-20 µM range. Not surprisingly second generation tetracyclines showed a higher affinity to the 30 S ribosome and the glycylglycine tigecycline and nanomolar KD values.
Crystal structures of the Thermus thermophilus ribosome in complex with tetracycline revealed that the primary binding site is located on the small 30S subunit [15, 16]. Tetracycline binds to the A site of the 30S subunit through interactions of ribosomal proteins with all four rings of the naphthalene core (Figure 3). These structures were the first direct visualization of the binding site for the drug. In each of the many structures solved since 2000, the primary binding site is always observed in the A site of the 30S ribosomal subunit at the interface of the mRNA and the entry point for amino-acyl tRNA molecules. This site sits on the base of the 30S subunit and interfere with the association of the larger 50S subunit needed to form the entire ribosome. Consequentially, translation of a mRNA molecule cannot commence and cell growth is effectively halted [4]. Thus, tetracyclines exert a bacteriostatic effect which prevents the spread of an infection until the innate immune system can clear the pathogen.
![Figure 3: Structure of the 30S ribosomal subunit of T. thermophilus in complex with tetracycline. Top: Ribbon diagram of the structure solved to a resolution of 3.4 Å. Bottom: Primary tetracycline binding site. The antibiotic is shown as a stick figure and binding involves interactions of the ribosome with all four rings of the naphthalene core. The figure was modified from reference [15].](/fulltextimages/8712/fig_3.png)
Figure 3: Structure of the 30S ribosomal subunit of T. thermophilus in complex with tetracycline. Top: Ribbon diagram of the structure solved to a resolution of 3.4 Å. Bottom: Primary tetracycline binding site. The antibiotic is shown as a stick figure and binding involves interactions of the ribosome with all four rings of the naphthalene core. The figure was modified from reference [15].
Tetracycline Resistance Mechanisms
The widespread use of tetracycline antibiotics over the past 70 years has led to the development of bacterial resistance in both clinical and agricultural settings. Tetracycline resistance began soon after it was first used and has been an increasing problem ever since [2, 4]. Resistance develops in three prevalent ways. One of these ways is efflux which is assisted by a group of membrane bound protein that pump the antibiotic out of the cell of the pathogen thereby preventing it from reaching the target ribosome. Another mechanism of tetracycline resistance involves ribosomal protection by which a series of proteins either physical block off the ribosome or work to dislodge the drug if it binds. The development of glycylcyclines has been particularly useful in overcoming ribosomal protection since substituents at C9 (Figure 2) stack with nucleobases of the ribosome increasing its affinity and preventing the binding of protection proteins [18]. Both efflux and ribosomal protection allow protein synthesis to occur in the pathogen even when tetracycline antibiotics are used for treatment of infection which has required the production of new generations of the drug every 20-30 years. A third and more recently discovered mechanism of tetracycline resistance involves the enzymatic oxidation of the drug which completely degrades it before it even enters the cell [3, 11, 12, 13, 19]. This section will briefly discuss each of the common ways tetracycline resistance has developed with an emphasis on enzymatic degradation that seems to be an emerging issue that is unlikely to be resolved through the production of novel tetracyclines.
Some of the earliest findings of resistance discovered a decrease in the amount of tetracycline found in cells bearing the plasmid of Escherichia coli R-factor R222 [2]. This was discovered during a cellular study of tetracycline treatment at Tufts University School of Medicine by Levy and McMurry. The R-factor used in the study represents the first demonstration of efflux and there are currently more than 20 known efflux proteins known in human pathogens [3, 10]. Efflux decreases the effective concentration of the drug in a bacterial cell since the proteins physically pump tetracyclines out of the cell and into the surrounding environment. Efflux was the first tetracycline resistance mechanism discovered and was reported as early as 1963 only a couple of decades after the first tetracycline was used clinically [20]. Efflux proteins are collectively referred to as “Tet” proteins and seven groups of efflux pumps have currently been identified [2, 3]. By lowering the effective concentration of tetracyclines in the cell Tet proteins decrease the probability of binding to the ribosome and shift the equilibria for binding towards free tetracycline and ribosome. This dramatically increases the amounts of protein that the pathogen is able to make to support its growth and survival.
Ribosomal protection by another group of Tet proteins are also a common way resistance to tetracyclines has developed. This mechanism was first found in streptococci and later in anaerobic bacteria [2, 3]. Ribosomal protection proteins physically block off the ribosome to prevent binding of the drug and to the 30S ribosomal unit and to promote the association of the 50S subunit to form the entire intact ribosome required for protein synthesis. The two most common ribosomal protection proteins are Tet(M) and Tet(O). These proteins cause tetracyclines to dislodge from the ribosome. They are GTPases with homology to elongation factors that bind to the ribosome analogously during translation. The dislodging of tetracyclines through the action of Tet(M) and Tet(O) dramatically reduces the effectiveness of the drug in clinical settings [5, 21]. Success has been achieved in overcoming resistance through this mechanism through the addition of bulky substituents at C9 of the naphthalene core seen in modern tetracyclines (Figure 2) as best studied in the case of tigecycline [7, 18].
A third mechanism of tetracycline resistance is chemical inactivation of the drug by flavin dependent monooxygenases [3, 11, 12]. The first such enzyme to be identified has been well characterized functionally, biochemically and structurally. This enzyme is named tetracycline monooxygenase (TetX) and has been classified as a group A flavin monooxygenase base on structural homology to other enzymes in this group, the type of electron donor a reduced nicotinamide adenine dinucleotide phosphate (NADPH) and the identity of the organic product of the reaction [22]. As shown in Figure 4, transforming recombinant E. coli cells with the gene encoding for TetX not only allowed for growth in the presence of tetracycline, but the culture media turned visibly dark [12]. As discussed in more detail below, the initial product of the TetX reaction is 11a- hydroxytetracycline which is unstable at physiological pH values and rapidly decomposes to a mixture of unidentified products that give rise to the darkening of the culture media. This would prevent the inhibition of protein synthesis by tetracycline in vivo since the reaction products cannot effectively bind to the ribosome. The environmental occurrence of tetracycline degrading enzyme is of great concern since horizontal gene transfer between soil microbes is common and already thought to be a source of the growing number of resistant bacteria in nature [23, 24].
Both first and second generation tetracyclines were very susceptible to efflux pump and ribosomal protection, but third generation antibiotics seem to have overcome these mechanisms for now [5, 21, 25, 26]. However, tigecycline is subject to degradation by TetX which was shown to effectively utilize the antibiotic as a substrate [11, 12]. Additional tetracycline degrading enzymes have been identified [3] and it does not appear that generating even newer versions of the drug will overcome this form of resistance. Instead, tetracycline degrading enzymes need to be studied in more detail to develop inhibitors to knock out their activities. These inhibitors could then be used in conjunction with glycylcyclines to treat infections caused by resistant bacteria.
![Figure 4: The effect of TetX expression on bacterial growth in presence of tetracycline. Liquid cultures of E. coli W3110, bearing the tetx2 gene on plasmid pDBI grown in the presence of tetracycline, turns the medium black which shows tetracycline degradation. The figure was modified from reference [12].](/fulltextimages/8712/fig_4.png)
Structural and Biochemical Studies on Tetracycline Monooxygenase
The physiological role of TetX has been definitively shown to confer tetracycline resistance to bacteria and several structures of the enzyme have been solved, to date, but only a limited number of kinetic or mechanistic studies have been carried out on the enzyme. This has largely resulted from the unavailability of a quick and easy expression and purification protocol for the enzyme needed to obtain large amounts of TetX for these studies. Such a protocol was recently described along with a polargraphic assay for TetX activity that measures the consumption of oxygen by the enzyme instead of the disappearance of NADPH during TetX turnover [19]. These results will make establishing the chemical mechanism of the TetX catalyzed reaction significantly more feasible. Once the chemical mechanism is known transition state analogues or mechanistic based inhibitors can be developed to prevent the degradation of tetracyclines by TetX. This review will end with a brief description of the expression and purification protocol recently developed as well as a summary of the biochemical and structural studies previously reported for the enzyme [13, 19].
Original reports of TetX described the cloning of the tetX2 gene encoding the enzyme into E. coli, and purification of the His6 tagged recombinant enzyme by nickel affinity chromatography [12]. The procedure resulted in 6 mg of TetX per 1 L of culture which was not only plenty of sample to establish the biochemical and structural properties of the enzyme (see below), but also provided the basis for an optimized protocol describe later [19]. The study reported in reference [19] began with the same recombinant plasmid harboring the tetX2 gene bearing a His6 tag, but used it to transform and screen a number of expression strains. The specific activities of cell extracts obtained from each strain was measured after induction of TetX expression at different times and temperatures through incubations with isopropyl β-D-1-thiogalactopyrande. It was found that induction of E. coli DH5α for 20 h at 30°C resulted in the highest expression level of TetX. Additionally, an ammonium sulfate precipitation step involving incubation of the cell extract with 40% saturation of salt was found to purify the sample without the need for additional chromatographic step beyond the nickel affinity column (although the study was not aimed to solve the structure of TetX which would require more highly pure samples). The published study described a yield of 36 mg/L of TetX and now routinely gives yields of up to 50 mg/L of culture.
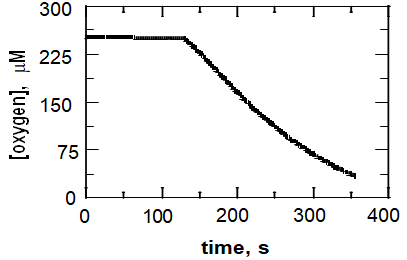
In addition to improving the amount of protein obtained, a quicker activity assay was described in reference [19] as compared to that used in previous reports of TetX [11, 12]. The kinetics of TetX was previously measured using the traditional spectrophotometric assay that monitors the oxidation of NADPH in the reductive half reaction of the enzyme, which takes between 10-15 minutes per substrate concentration used. Instead, the assay developed in [19] utilizes a Clarke-type oxygen electrode to monitor the consumption of oxygen occurring during the oxidative half reaction of TetX. A typical oxygen electrode trace is shown in Figure 5. As shown in the trace the activity assay only takes ~4 minutes allowing for a quicker determination of the steady state kinetic parameters of the enzyme. This will allow for detailed kinetic experiments in which tetracycline, NADPH and oxygen are varied which will establish the steady state kinetic mechanism of the enzyme to provide a framework to elucidate the chemical mechanism of TetX.
The work will build on previous biochemical studies of the enzyme carried out by the group of Gerard Wright at McMaster university [11, 12]. These studies demonstrated that TetX requires NADPH which cannot be substituted by NADH and that oxygen is essential for activity. TetX catalyzes the inactivation of the broad- spectrum tetracycline antibiotics. These include each of the first and second generation tetracyclines listed in Figure 2, with no obvious preferred substrate under the conditions tested (pH 8.5 and room temperature). The specifity constants were between 103-104 M-1s-1 for each of the first and second generation tetracyclines tested and the turnover numbers were also similar (~0.3-1 s-1) [12]. The clear demonstration of TetX inactivation of first and second generation tetracyclines demonstrate that chemists will not win the battle against pathogens by overcoming efflux and ribosomal protection alone. Furthermore, it was later shown that TetX can also degrade glycylcyclines such as tigecycline [11] rising a serious concern that even the latest tetracyclines to reach the market may soon prove ineffective if TetX activity becomes widespread in clinical and agricultural settings.
Wright et al were able to carefully isolate the product of the TetX reaction at low pH to determine its chemical structure [12]. By using mass spectrometry, 1H and 13C NMR the reaction product of TetX was shown to be 11a-hydroxytetracycline. The reaction scheme of the enzyme is thus that shown in Scheme 1. Considerable effort had to be exerted to obtain a stable product to for structure determination. The instability of the TetX product at physiological pH is presumably what gives rise to the dark color of the liquid media shown in Figure 4. The
exact identity of the polymer(s) that darkened the culture was not determined but it is likely a complex mixture of decomposition products of 11a-hydroxytetracycline. Importantly no competes of this mixture can bind to the ribosome to inhibit protein synthesis and the E. coli culture was able to live and grow as normal even in the presence of the antibiotic. Despite the discovery of the reaction product by Wright et al no information concerning the timing of bond cleavage and formation events during the TetX reaction are known. These will be needed to generate mechanism based inhibitors of TetX to aid in the fight against tetracycline resistance.
![Figure 6: Overall Structure of TetX indicating the sub-division into two domains binding a FAD cofactor. The figure is from [13].](/fulltextimages/8712/fig_6.jpeg)
Scheme 1: The reaction catalyzed by TetX. The oxygen atom incorporated into the produced is highlighted in red. NADPH = reduced nicotinamide adenine dinucleotide phosphate. The fight will be aided by structural studies of TetX that have also been published [13, 27]. TetX is a monomer comprised of two major domains binding a single FAD molecule (Figure 6). The structure of the enzyme was solved in the absence of tetracycline substrates and with the substrate analog 7-iodotetracycline to resolutions of 2.1 and 2.4 Å, respectively [13]. The structural map provided by Volker et al provides a platform for detailed biochemical studies of the enzyme in which active site amino acid can be changed using site-directed mutagenesis as a tool. It also shows the typical Rossman fold and structural homology to group A flavin monooxygenases (FMOs) [22]. This classification provides a working model for the potential catalytic mechanism based on other group A FMOs as described below. However, in order to rationally design mechanism based inhibitors of TetX to fight tetracycline resistance greater details on the precise timing of bond cleavage and formation events as well as identification of the rate-limiting step(s) of the reaction need to be identified.
The overall oxygenation mechanisms for group A FMOs is shown in Scheme 2 [22, 28]. In the reductive half reaction (higlighted in yellow), a hydride transfer between a reduced NAD(P)H substrate and an enzyme bound FAD cofactor generates the hydroquinone form of flavin (reduced flavin) and NAD(P)+. In the oxidative half reaction (highlighted in blue), the reduced flavin reacts with molecular oxygen to form a C4(a)- peroxyflavin (peroxyflavin), which either directly oxygenates the organic substrate of the enzyme (Path A) or is protonated to form a hydroperoxyflavin, which then incorporates a single oxygen atom into the substrate (Path B). The catalytic cycle is then completed through expulsion of water from the hydroxyflavin form of the enzymes to regenerate the fully oxidized cofactor of the resting enzyme.
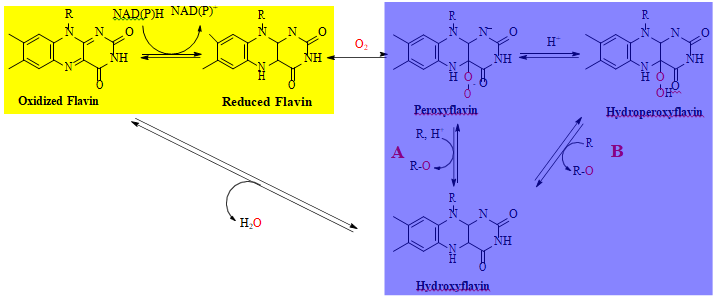
Scheme 2: General Mechanisms of Groups A FMOs highlighting the reductive (yellow) and oxidative (blue) half reactions. The oxygen atom that is incorporated into the organic product is highlighted in red and R is the organic component of the reactant or product (tetracycline and 11a-hydroxyteracycline in the v=case of TetX.
Conclusion
The modifications at the C9 position of the naphthalene core of tetracyclines made to second generation antibiotics to produce glycylcyclines not only greatly increased the affinity of the drug to the target ribosome, but dramatically protected it from both efflux and ribosomal protection mechanisms employed by pathogenic bacteria. This has proven to be an instrumental weapon for medicinal chemists in the battle against pathogenic bacteria. However, the fight is not over and it seems bacteria may be regrouping and starting to employ new weapons of their own to survive in the presence of tetracycline antibiotics. The weapons involve the production of tetracycline inactivating enzymes such as TetX. The enzyme hydroxylates tetracyclines of each generation to produce a completely unstable product that is unable to bind to the ribosome to prevent bacterial protein synthesis. The demonstration of TetX activity with even the glycylcycline tigercycline demonstrates that the battle plan employed by medicinal chemists needs modifications. In addition to fighting efflux and ribosomal protection which history has shown bacteria evolve new proteins for these mechanisms every 20-30 years, inhibitors of tetracycline degrading enzymes must also be developed. These inhibitors will reinforce new generation tetracyclines and be used in conjugation with the antibiotics. By knocking out TetX activity the tetracyclines will be protected and can bind to the bacterial ribosome to stop protein synthesis. This will prevent cellular growth and allow the innate immune system to clear the pathogen and treat infections.
Acknowledgements
This work was supported by a Departmental Research Grant from The Robert A. Welch Foundation AC- 0006 and the Ronald E. McNair Program (to K.A.).
References
-
Chopra I, Roberts M (2001) Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65(2): 232-260.
-
Nelson ML, Levy SB (2011) The history of the tetracyclines. Ann N Y Acad Sci 1241: 17-32.
-
Markley JL, Wencewicz TA (2018) Tetracycline- Inactivating Enzymes. Front Microbiol 9: 1058.
-
Nguyen F, Starosta AL, Arenz S, Sohmen D, Donhofer A, et al. (2014) Tetracycline antibiotics and resistance mechanisms. Biol Chem 395(5): 559-575.
-
Petkovic H, Lukezic T, Suskovic J (2017) Biosynthesis of Oxytetracycline by Streptomyces rimosus: Past, Present and Future Directions in the Developmentof Tetracycline Antibiotics. Food Technol Biotechnol 55(1): 3-13.
-
Noskin GA (2005) Tigecycline: a new glycylcycline for treatment of serious infections. Clin Infect Dis 41(5): 303-314.
-
Stein GE, Babinchak T (2013) Tigecycline: an update. Diagn Microbiol Infect Dis 75(4): 331-336.
-
Granados Chinchilla FC, Rodriguez C (2017) Tetracyclines in Food and Feedingstuffs: From Regulation to Analytical Methods, Bacterial Resistance, and Environmental and Health Implications. J Anal Methods Chem 2017: 1315497.
-
Roberts MC (1996) Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol Rev 19(1): 1-24.
-
Thaker M, Spanogiannopoulos P, Wright GD (2010) The tetracycline resistome. Cell Mol Life Sci 67(3): 419-431.
-
Moore IF, Hughes DW, Wright GD (2005) Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry 44(35): 11829-11835.
-
Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, et al. (2004) TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem 279(50): 52346-52352.
-
Volkers G, Palm GJ, Weiss MS, Wright GD, Hinrichs W (2011) Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett 585(7): 1061-1066.
-
Olson MW, Ruzin A, Feyfant E, Rush TS, JO Connell, et al. (2006) Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob Agents Chemother 50(6): 2156-2166.
-
Brodersen DF, Clemons WM, Carter AP, Morgan Warren RJ, Wimberly BT, et al. (2000) The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103(7): 1143-1154.
-
Pioletti M, Schlünzen F, Harms J, Zarivach R, Glühmann M, et al. (2001) Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J 20(8): 1829-1839.
-
Aleksandrov A, Simonson T (2008) Molecular dynamics simulations of the 30S ribosomal subunit reveal a preferred tetracycline binding site. J Am Chem Soc 130(4): 1114-1115.
-
Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, et al. (2013) Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Nat Acad Sci 110(10): 3812-3816.
-
Bernal J, Ojutiku Z, Cordero A, Martinez A, Berkey K, et al. (2020) An Improved Protocol for the Expression and Purification of Tetracycline Monoozyhenase: An Enzyme Involved in Antibiotic Resistance. Annals of Advanced Biomedical Science 3(2): 153-160.
-
Izaki K, Arima K (1963) Disapperance of Oxytetracycline Accumulation in the Cells of Multiple Drug-Resistant Esherichoa coli Nature 200: 384-385.
-
Connell SR, Tracz DM, Nierhaus KH, Taylor DE (2003) Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother 47(12): 3675-3681.
-
Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ (2014) Flavin dependent monooxygenases. Arch Biochem Biophys 544: 2-17.
-
Du B, Yang Q, Wang R, Wang R, Wang Q, et al. (2019) Evolution of Antibiotic Resistance and the Relationship between the Antibiotic Resistance Genes and Microbial Compositions under Long-Term Exposure to Tetracycline and Sulfamethoxazole. Int J Environ Res Public Health 16(23): 245-251.
-
Duan M, Gu J, Wang X, Li Y, Zhang R, et al. (2019) Factors that affect the occurrence and distribution of antibiotic resistance genes in soils from livestock and poultry farms. Ecotoxicol Environ Saf 180: 114-122.
-
Liu Y, Jia Y, Yang K, Li R, Xiao X, et al. (2020) Anti-HIV agent azidothymidine decreases Tet(X)-mediated bacterial resistance to tigecycline in Escherichia coli. Commun Biol 3(1): 162.
-
Pankey GA (2005) Tigecycline. J Antimicrob Chemother 56(3): 470-480.
-
Volkers G, Damas JM, Palm GL, Panjikar S, Soares CM, et al. (2013) Putative dioxygen-binding sites and recognition of tigecycline and minocycline in the tetracycline- degrading monooxygenase TetX. Acta Crystallogr D Biol Crystallogr 69(9): 1758-1767.
-
van Berkel WJ, Kamerbeek NM, Fraaije MW (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol 124(4): 670-689.
- Origin, Evolution, and Functional Impact of Short Insertion- Deletion Variants in Human Genomes: A Review
- Harnessing Molecular Glues for Next-Generation Vaccine, Cancer and Cardiovascular Disease Drug Development: A Comprehensive Review
- Lateral Cervical Epidermal Inclusion Cyst in a Paediatric Patient: A Rare Case Report
- Malarial Plasmodium Falciparum with Hepatitis B and C Virus Infections among Blood Donors in Ife Central Local Government Area, Ile Ife, Osun State, Nigeria
- Withanolides and Withaferin A- What’s next in Ashwagandha Research
- Designing of Dual Pulse Photoacoustic Tomography for Imaging of Drug-Response and Tumor Growth