Posterior Reversible Encephalopathy Syndrome in a 12-Year- Old Boy with Mixed Connective Tissue Disease
Reversible posterior encephalopathy syndrome is a clinic radiologic entity characterized by headaches, altered mental status, seizures, and visual loss and is associated with white matter vasogenic edema predominantly affecting the posterior occipital and parietal lobes of the brain. We herein, present a rare complication of mixed connective tissue disease. A 12-year old boy presented with generalized tonic clonic fits, aphasia, fever and generalized body swelling since 2 weeks. His labs showed hypercholestremia, hypoalbuminemia and massive proteinuria. Renal biopsy revealed secondary membranous glomerulopathyand and he developed posterior reversible encephalopathy syndrome. His anti RNP and anti-smooth muscle came back positive. Evaluation by magnetic resonance scan of the brain showed that the child had hyper intense signal in the parieto-occipital regions. All signs of PRES started resolving after seven days of supportive treatment evidenced by subsequent radiological evaluation. PRES and secondary membranous glomerulopathyshould be kept in mind in any connective tissue disease that is on prolonged immunosuppressive therapy.
Introduction
Posterior reversible encephalopathy syndrome, a rare disorder of central nervous system, was first described by Hinchey in 1996. It was defined as a variable combination of consciousness impairment, seizure activity, headaches, vomiting, visual abnormalities (hemianopia or cortical blindness) and focal neurological signs, which was associated with neuroimaging abnormalities characterized by partially or completely reversible subcortical vasogenic edema in the posterior white matter [1]. PRES can develop in association with a vast array of clinical conditions including autoimmune diseases (such as SLE or Wegener granulomatosis), systemic infections, pre-eclampsia, hypertension, organ transplantation, malignancies, chemotherapy, and immunosuppression [2]. Here, we present a case of PRES in a 12-year-old boy with steroid-resistant membranous glomerulopathyand mixed connective tissue disease [3].
Case Report
A 12-year-old boy presented with generalized tonic clonic fits, aphasia, fever and generalized body edema, proteinuria of 3318.9mg/24 hr, low serum albumin of 1.87 g/L, high serum cholesterol of 465 mg/dland initial arterial blood pressure of 109/71 mmHg. The past history suggested the patient having Reynaud’s phenomenon 9 months ago for which he received monthly IM injections – benzyl penicillin and steroids. He was admitted in Kabul for the complaint of generalized body swelling. He was suspected minimal change disease for which he received deltacortil (3+3+3) for one and a half month. The workup showed nothing significant and the steroids were stopped. The child developed sudden headache, generalized tonic clinic convulsion and developed aphasia.
Magnetic resonance venography was done on 11th February showed bilateral cerebral abnormal signal foci with surrounding edema and no enhancement. Complementary CT did not reveal any hemorrhages. MR showed hyper intense signal in the parieto-occipital regions, and axial FLAIR images and DWI revealed bilateral cortical and subcortical white matter edema in parieto-occipital lobes, and it showed symmetrical hypo densities in parieto-occipital regions of cerebral hemispheres (Figure 1). He was diagnosed with posterior reversible encephalopathy syndrome.
On arrival at Northwest General Hospital, his blood pressure was 110/70 mmHg, pulse 99 beats per minute and temperature 36.8°C. Neurological examination showed normal cranial nerve function, equal and symmetric deep tendon reflexes. Muscle strength was 4/5 in bilateral upper and lower extremities.
On laboratory evaluation, the child had no evidence of infection. He had massive urinary protein excretion of 138 mg/kg/24 hr, low serum albumin of 1.87 g/L, normal serum urea of 22mmol/L, normal serum creatinine of 0.23mg/dl and normal serum electrolytes levels. The LP report was insignificant.
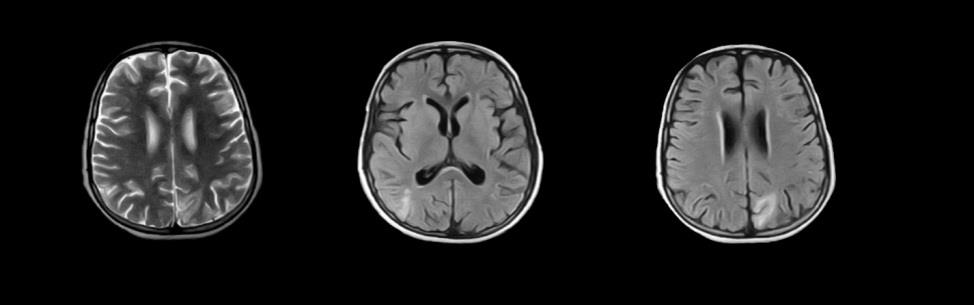
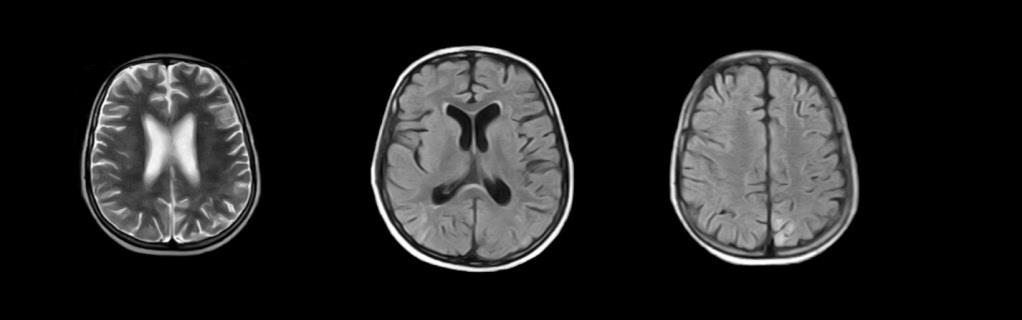
He was found to have steroid-resistant nephrotic syndrome and his kidney biopsy revealed secondary membranous glomerulopathy. The boy was treated with steroids and cyclosporine. On day sixth of hospitalization his symptoms improved but he was still mute. Follow-up MRI after seizures demonstrated mild resolution of the initial cerebral lesions in comparison with previous MR done on 11-02-2017 (Figure 2). The MR report confirmed the patient to have PRES. Keeping in check the secondary glomerulonephritis status, Anti-DsDna came negative and ENA profile was done. The boy turned out having Anti RNP and Anti smooth muscle positive. The child was treated with anti-epileptic, diuretic, steroids and immunosuppressant (cyclosporine A), thereafter he got no seizure and showed improvement. His blood pressure was kept at 120/70 mmHg and urinary protein excretion gradually decreased. After 20 days in the hospital, he was discharged home on tapering steroids, anti-epileptics and diuretics.
Discussion
PRES was first described by Hinchey et al in 1996. Over the past decade, with advance in neuroimaging techniques, PRES has been shown to be associated with mixed connective tissue disease, nephritic syndrome, Henoch-Schönlein purpura, post streptococcal glomerulonephritis, hemolytic uremic syndrome, hypertension, Addison’s disease, ganglion-neuroma, acute lymphoblastic leukemia, intra-abdominal neurogenic tumors, porphyria, bone marrow transplant and steroids in children [4, 5]. Clinical Features The typical features of PRES consist of consciousness impairment, seizure activity, headaches, visual abnormalities, nausea/vomiting, and focal neurological signs (Table 1). This mixed connective tissue disorder case presented with headache, seizures, blindness, confusion and hypertension, with cyclosporine a treatment.
Clinical Features of PRESS
Consciousness
impairment Seizure activity
Headaches Visual abnormalities
Nausea/vomiting Focal neurological signs
Acute hypertension
| Radiological Features of PRESS |
| Bilateral |
| Asymmetric |
| Confluent |
| Gray matter |
| Posterior > anterior |
| Occipital |
| Parietal |
| Frontal |
| Temporal |
| Brainstem |
| Cerebellum |
| Basal ganglia |
Table 1: Features of PRES.
The exact pathophysiological mechanism of PRES remains unclear. Three hypotheses have been suggested, which include: i. After exposure to causative agent (such as severe hypertension), auto regulation mechanism of intracranial pressure fails, leading to vasogenic edema. ii. After exposure to causative agent (such as mild-to- moderate hypertension), cerebral vasoconstriction and hypo-perfusion cause vasogenic brain edema and ischemia. iii. Endothelial injury with disruption of the blood-brain barrier leads to fluid and protein transudation in the brain [6]. iv. In addition, immunosuppressive agents such as methylprednisolone, dexamethasone, cyclosporine and cyclophosphamide have been reported to be related to PRES [7, 8].
It’s speculated that, in this case, a positive underlying cause of mixed connective tissue disease, changes in blood pressure, severe hypoalbuminemia and cyclosporine A administration may be the candidate causes of PRES. Diagnosis PRES can be diagnosed according to typical clinical manifestation and neurological image examination. Patients are generally presented with headache, vomiting, visual perception abnormalities and seizures, and radio logically characterized by symmetric distribution of patchy or globe-like lesions in the white matter of the parietal and occipital lobes. CT scan, which is usually easier to perform first, shows multiple hypo densities in the cortical-subcortical areas, which is different from acute infarct or hemorrhage. On magnetic resonance imaging, T1-weighted hypointense, T2-weighted hypointense and T2-weighted FLAIR hypointense areas are bilaterally revealed in the occipital and parietal lobes, which can partially or completely resolved on follow-up scans (Figure 3). In general, vasogenic edema is considered to account for the pathophysiology of PRES, but the presence of cytotoxic edema is the main prognostic factor for the condition as it may signify irreversible brain injury. DWI sequence can be helpful in differentiating between cytotoxic edema and vasogenic edema. In our patient, hypointense signal in the parieto- occipital regions, and axial FLAIR images and DWI revealed bilateral cortical and subcortical white matter edema in parieto-occipital lobes, and it showed sym- metrical hypo densities in parieto-occipital regions of cerebral hemispheres.
Treatment
PRES must be managed carefully; the pathogenic factors should be clarified if possible since remove of etiologies is very important for the successful treatment.
In hypertension- related and drug-induced PRES, effective management includes prompt withdrawal of offending agent, aggressive control of blood pressure, timely anti- convulsion. Controversy still exists whether immunosuppression should be continued in the treatment of PRES with steroid-resistant nephritic syndrome. In our case, hypertension was undoubtedly an important cause, but we were uncertain whether cyclosporine A also played a pathogenic role. Considering that extreme hypoalbuminemia may worsen brain edema, and patient could not get remission of nephritic syndrome without powerful immunosuppression treatment, we continued cyclosporine A and steroid treatment along with prompt anti-hypertension and anti-seizure, and the patient gradually recovered of not only PRES but also nephritic syndrome. It suggests that immunosuppressive agents can be cautiously administrated in membranous glomerulopathywith PRES.
A similar case report was found in Iran. Most PRES cases were fully reversible in a matter of days to weeks, with timely treatment. However, prolonged seizures and hypertension may result in death or permanent neurological disability. A 15-year old Iranian boy was diagnosed with mixed connective tissue disease, and cyclophosphamide pulse therapy was administration. After 8 days, the brain magnetic imaging abnormalities were resolved following the antihypertensive and antiepileptic treatment. The observations had indicated posterior eversible encephalopathy. The patient developed respiratory distress and eventually died [9]. Therefore, prompt recognition and timely rational management is important to prevent irreversible neurological deficits.
Our patient presented with PRES along with the features of membranous glomerulopathylead. Therefore, initially, we could not accurately determine the main causative source of PRES among the cyclosporine therapy, steroid, hypertension, nephritic syndrome and the flare- up of the underlying disease. Most of the reported cases of PRES are suggested to be due to cytotoxic or steroid therapy. In the present case, the diagnosis of PRES and the biopsy revealing secondary glomerulopathylead us to believe that there is another potential cause and it was confirmed by the positive ENA profile. As in our case, PRES and Nephritic were concomitant with mixed connective tissue disease. Therefore, PRES may be a manifestation of mixed connective tissue disease in our case because our patient presented PRES during the disease flare-up.
To the best of our knowledge, a case of PRES in a patient with mixed connective tissue disease has only been reported once in Iran in 2016. The manifestations of it have been believed to be less frequent than findings of other systems [9]. Although the main neurological manifestations of mixed connective tissue disease are trigeminal neuropathy, headaches, and aseptic meningitis, this report suggests PRES as a neurological condition which may occur during the course of the disease. Although there is no difference between our patient and previously reported cases in terms of PRES characteristics, further reports are needed for better understanding of PRES in mixed connective tissue disease.
Conclusion
Through our case report, we wish to highlight that mixed connective tissue disease along with membranous glomerulopathyshould be considered risk factors for developing PRES in children. An awareness of this observation is crucial for timely diagnosis and treatment, and therefore minimizing the risk of permanent neurologic deficits.
Cases of PRES have been widely reported since its first description and various factors such as etiology have been identified; however, numerous aspects regarding the pathogenesis of this entity are yet to be elucidated as in this case. The treatment of PRES, as a secondary pathology, depends upon the determination of the underlying contributing condition; however, palliative therapy for symptoms that might worsen the outcome (e.g. seizures) must be provided, as well as strictly monitored BP control.
References
-
Hinchey J, Chaves C, Appignani B, Breen J, Pao L, et al. (1996) A reversible posterior leukoencephalopathy syndrome. N Engl J Med 334(8): 494-500.
-
Lee VH, Wijdicks EF, Manno EM, Rabinstein AA (2008) Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol 65(2): 205-210.
-
Fugate JE, Claassen DO, Cloft HJ, Kallmes DF, Kozak OS, et al. (2010) Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc 85(5): 427-432.
-
Kwon S, Koo J, Lee S (2001) Clinical spectrum of re- versible posterior leucoencephalopathy syndrome. Pediatric Neurol 24(5): 361-364.
-
Prasad N, Gulati S, Gupta RK, Sharma K, Gulati K, et al. (2007) Spectrum of radiological changes in hypertensive children with reversible posterior leucoencephalopathy. Br J Radiol 80(954): 422-429.
-
Bartynski WS (2008) Posterior reversible encepha- lopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol 29(6): 1043-1049.
-
Kim JS, Lee KS, Lim SC, Ahn JY, Song IU, et al. (2007) Reversible posterior leukoencephalopathy syndrome in a patient with multiple system atrophy: a possible association with oral midodrine treatment. Mov Disord 22(7): 1043-1046.
-
Pinedo DM, Shah-Khan F, Shah PC (2007) Reversible posterior leukoencephalopathy syndrome associated with oxaliplatin. J Clin Oncol 25(33): 5320-5321.
-
Reza R, Rammin R, Mozhdeh Z (2016) Posterior reversible encephalopathy syndrome in a patient with mixed connective tissue disease: A case report. J med case rep 10(1): 145.
- Are the Vaccines the Only Solution to Prevent the COVID-19 Pandemic? Part Two
- Clinical Characteristics of Women in this New Global Immunodeficiency
- Cell Dynamics in HIV Pathogenesis: Insights and Implications
- Determination of the CDR (CDR1, CDR2) « Complementary- Determining Region Invertebrate Primitive Antibody from Sea Star »
- Prioritizing Care for High-Risk COVID-19 Patients in the EU: 10 Civic Recommendations to the Institutions
- Comprehensive Insights into ModRNA Vaccines: Persistent PP-Spike Recombinant Protein, Hyperimmune/Inflammatory Reactions, Thrombotic Vasculopathy, Chronic Organ Complications and Excess Deaths