DAMPs and Gliosis
Glial cells change their morphology, molecular signature, and functional events in response to neurodegenerative diseases and injury in the brain and retina. This process is termed gliosis. Gliosis is context-dependent, based on the severity of the diseases or damage, and can be protective or detrimental to neural functioning. Recent reports show that Damage-Associated Molecular Patterns (DAMPs) are endogenous danger molecules that are released from damaged or dying cells associated with gliosis. This article is focused on the pathological and protective role of extracellular, cytosolic, and nuclear DAMPs on gliosis.
Introduction
Gliosis is the universal reaction by glia to the injury or disease conditions in the brain and retina for sealing off the injured tissue, promoting tissue integrity, and restricting inflammation and neuronal death [1, 2]. Major glial cells that take part in this process are astrocytes, NG2-expressing glial precursors, and microglia in the brain, and astrocytes, Muller glia, and microglia in the retina [3, 4, 5]. It is regulated by a large number of extracellular signals generated after brain injury and retinal diseases. These molecules include damage-associated molecular patterns (DAMPs) released by the damaged or dead cells; molecules entering via leaky blood-brain barrier (BBB) or blood-retinal barrier (BRB), molecules released by infiltrating leukocytes, and molecules released by reactive glia themselves [3, 6, 7]. The severity of the gliosis depends upon the quantity, type or combination, and duration of these molecules [3]. Gliosis is a part of coordinated multicellular innate and adaptive immune responses to brain injury and retinal diseases.
DAPMs in Gliosis
DAMPs are endogenous danger molecules released from the extracellular and intracellular space of the damaged tissue or dead cells, which can capable of triggering and perpetuating an immune response in the body. DAMPs can interact with cells through multiple signaling pathways such as the RAGE pathway, TLR-pathway, NLRP3 inflammasome pathway, CD-14 dependent pathway, IL-R1/ST-2 signaling pathway, CD-91 signaling pathway, and DNGR-1 signaling pathway [6, 7]. DAMPs could be similarly acting with glia as the pathways are not deciphered in detail. The reactive glia activated by DAMPs can further secret cytokines and chemokines and play a significant role during inflammation. Reactive Muller glia play important role in neuroinflammation, neurodegeneration, and vascular permeability by secreting harmful factors, such as tumor necrosis factor (TNF), and interleukin-1 (IL-1), nitric oxide (NO), ATP, Histones H3, and H4. The extracellular ATP and histones further act as DAMP and activate P2X7R and TLR respectively leading to neuroinflammation and neurodegeneration in retinal diseases [2, 7, 8]. Whereas, the inflammatory markers may activate other glial cells such as astrocytes and microglia generating a vicious cycle of inflammation and may play role in neurovascular damage in the retina [8]. Activated microglia/ macrophage and astrocytes secret pro-inflammatory factors including TNF-α, IL-6, IL-1α, IL-1β, CCL-2, CCL-3, CCL-4, CCL- 5, CXCL-1, CXCl-2, CXCL-10, NO, and play important role in neuroinflammation and degeneration, leukocyte infiltration, and neuro-vascular damage in neuronal and retinal diseases [4, 9, 10, 11].
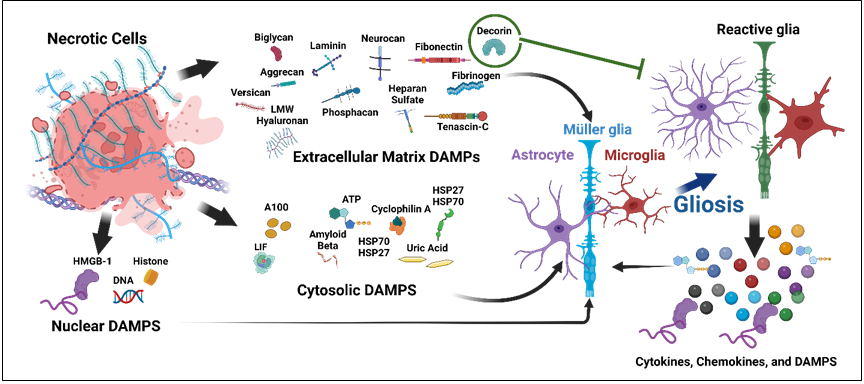
Figure1: The schematic representation of DAMPs in gliosis. The injured neuronal tissue and diseased retina produce extracellular, cytosolic, and nuclear DAMPs, which activate glia (astrocyte, Muller glia, and microglia). The reactive glia produces cytokines, chemokines, and DAMPs which can again activate glia to create a vicious cycle of neuroinflammation, except decorin, an extracellular DAMPs which shows inhibitory effect on gliosis.
Extracellular DMPs and Gliosis
The extracellular DAMPs that play a pathological or protective role in neuronal and retinal diseases are decorin, biglycan, versican, aggrecan, phospacan, neurocan, LMW hyaluronan, heparan sulfate, fibronectin, fibrinogen, laminin, and tenascin-c [6, 7]. Decorin is a small, leucine- rich proteoglycan, that plays a protective role in retinal and neuronal diseases [7, 12]. It significantly inhibits gliotic scar by acting on TGF-β in penetrating incisional wounds of the rat brain and juvenile post-hemorrhagic communicating hydrocephalus, as well as by inhibiting GFAP positive astrocytes, laminin, fibronectin, and endothelin-1 expression and microglia activation in the brain [12, 13]. TGF-β is strongly associated with astrogliosis, neuronal damage, and pathogenesis of glaucoma. It is reported that decorin is secreted by astrocytes naturally and the treatment of decorin inhibits the secretion of TGF-βs and connective tissue growth factor in human optic nerve astrocyte and optic nerve and further reduces the expression of collagen and fibronectin and play important role in the expression of ECM molecules. It may be useful as a therapeutic for glaucoma [14]. Biglycan, a small, leucine-rich chondroitin sulfate proteoglycan associated with retinal and neuronal diseases, though its role in disease pathology and gliosis is not known [7, 15]. It is reported that the biglycan expression significantly increases at the sight of injury, especially in the extracellular space and ED-1 positive cells [16]. Chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic central nervous system glial scar, which indicated that phosphacan and neurocan are in areas of gliosis may contribute to axonal regenerative failure after central nervous system (CNS) injury [17]. In contrast, brevican and versican are not expressed in the chronic glial scar [17]. In spinal cord injury, all the chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are significantly increased in the injury site with co-localization of neurocan with GFAP indicating reactive astrocyte as its source [18]. Also, the expression of versican is significantly increased in reactive Muller glia of light- induced retina degeneration model and advanced stage of retina degeneration Rho-/-mouse model [2, 19]. Astrocyte is the source of aggrecan that plays important role in the inhibition of neurite growth [20]. Also, the overexpression of aggrecan in multiple sclerosis lesions and gliotic retina of cadaveric proliferative vitreoretinopathy human eye is reported [10, 21]. Aggrecan could play a regulatory role in the activation of astrocytes. On the other hand, the microglia not only co-localize with a perineuronal net but also secret protease to degrade ECM molecules like aggrecan [22]. Hyaluronan is a major glycosaminoglycan of the extracellular matrix, and may play a pathologic or protective function based on its molecular weight. The LMW hyaluronan enhances astrocyte proliferation and gliosis and neuroinflammation whereas HMW hyaluronan inhibits glial scar formation and neuroinflammation in the brain and spinal cord injury [23, 24]. It also involves liquefication of vitreous in diabetic retinopathy and retinal detachment [7]. However, its role in Muller gliosis and microglial activation is unknown. Heparan sulfate proteoglycans play important role in cell adhesion, as a binding site for proteases, and regulation of growth factors such as basic fibroblast growth factor. Activated microglia releases cathepsin S which in turn degrades heparan sulfate proteoglycans by releasing heparan sulfate that could act as a DAMP [25].
On the other hand, heparan sulfate may mediate its regulatory role in various retinal and neuronal diseases like age-related macular degeneration, glaucoma, and diabetic retinopathy by interacting with various angiogenic growth factors, including FGF, VEGF, TNF-a, TGF-b, and IFN-g [26]. In an injured brain and TGF-β treated cultured glia, the sulfated heparan sulfate proteoglycan and syndecan-1 are significantly increased in astrocytes, suggesting novel approaches to modulating scar formation [27]. Fibronectins are the glycoproteins involved in different cellular functions including cell adhesion, chemotaxis, and opsonization. The deposition of fibronectin and fibrinogen is significantly increased in MS lesions and inflammatory and non-inflammatory disorders of CNS [28]. Fibronectin is synthesized by the astrocytes in normal conditions, and the activated astrocyte is associated with fibronectin, laminin, and collagen deposition during spinal cord injury [29, 30]. The role of fibronectin in microglia activation has been studied in a brain cryoinjury model in rats, and found that at the injury site there is a significant increase in fibronectin as well as activated microglia, macrophage, and astrocytes. Interestingly, the fibronectin is co-localized with the ED-1 positive microglia. The results suggest that fibronectin is extravasated into injured brain lesions via an impaired blood-brain barrier and stimulated microglia/macrophage activation, which modulates CNS inflammation after brain injury [31].
The idiopathic epiretinal membrane, a vision-threatening condition, is termed epiretinal gliosis characterized by reactive Müller cell gliosis. Under TGF-β treatment the Muller glia undergoes epithelial to mesenchymal transition with a significant increase in fibronectin gene expression and extracellular fibronectin deposition [32]. More interestingly, fibrinogen can also activate astrogliosis by promoting the availability of active TGF-beta after vascular damage in central nervous system injury [33]. The fibrinogen can induce a rapid and sustained microglial response, microglia clustering which is necessary for the development of axonal damage in neuroinflammation in multiple sclerosis [34]. However, the role of fibrinogen for Muller glia activation and vice versa is not known. Laminin expression significantly increases at the site of neuronal injury after astrogliosis and seems to have beneficial to CNS repair. Laminin promotes axonal growth, and laminin up-regulation after CNS injury has been correlated with the transformation of ramified, bipolar/rod- shaped microglia into amoeboid microglia [35, 36]. The role of laminin on Muller gliosis is not known, however, Laminin-1 dysregulation results in retinal Muller glia cell migration. Tenascin-C immunoreactivity was significantly higher with the reactive astrogliosis in the hippocampus of epileptic rats [37]. The loss of extracellular Tenascin-C lead to loss in gliosis in an autoimmune glaucoma mouse model [38]. Also, the activation of Tlr4 by Tenascin-C regulates chemotaxis, phagocytosis, and proinflammatory cytokine production in microglia [39]. Overall, the extracellular matrix DAMPs are related to gliosis and the mechanism of action could be studied in detail.
Cytosolic DAMPs and Gliosis
The cytosolic proteins such as leukemia inhibitory factor (LIF), S100 proteins, uric acid, heat-shock proteins (HSP), adenosine triphosphate (ATP), cyclophilin A, and amyloid-β (Aβ) play a pathological or protective role in retinal and neuronal diseases [6, 7, 11, 40, 41]. LIF is a pleiotropic cytokine and a member of the GP-130 class of neuropoietic cytokines. It is significantly increased in the brain and spinal cord injury [42]. The up-regulation of LIF is associated with astrocyte and Muller gliosis and activation of microglia, survival, and regeneration of injured peripheral neurons. LIF is also the key mediator of inflammatory responses in the peripheral, central nervous, and spinal cord injury systems [5, 40, 42]. The S100 proteins, a family of calcium-binding cytosolic proteins plays dual effects on the neuron, pro-survival at nanomolar concentration and inflammatory effects at higher concentrations [43]. The chronic over-expression of S100B leads to both astrogliosis and microgliosis in the brain during Alzheimer’s disease and is involved in the pathological progression of the disease by promoting amyloidogenic amyloid precursor protein processing [11]. The pharmacological inhibition of S100B shows inhibition of gliosis and neuronal loss in the Alzheimer’s disease mouse model [43]. Interestingly S100 alone or S100 and HSP27 lead to retinal gliosis and ganglionic cell loss in the autoimmune glaucoma model [44]. Uric acid (UA), an end product of purine metabolism, induces astrogliosis, microgliosis, inflammation, and neurodegeneration in rat hippocampus, by activating TLR4/NFkB signaling pathway [45]. NFkB inhibitor and TLR-4 knock-out mice show significant amelioration of UA-induced hippocampal inflammation and memory deficits in the rodent’s model. In diabetic patients and STZ-induced rats, monosodium urate is increased significantly as a byproduct of UA. Hypouricemic drugs allopurinol treatment significantly inhibits gliosis in STZ-induced DR rat retina, with inhibition of NLRP3, TLR-4, and IL-1β expression indicating involvement of UA in retinal gliosis via NLRP-3 signaling pathway. Heat-shock proteins (HSPs) are highly conserved “stress proteins.” HSPs are well known as intracellular molecular chaperones and also participate in morphological development and apoptotic processes [46].
The HSPs such as hsp27 and αβ-crystallin is highly expressed by the activated astrocytes and αβ-crystallin in activated microglia in Alzheimer’s disease and Parkinson’s disease, and are strongly associated with neurofibrillary degeneration and dementia [41]. When HSP90, HSP70, and HSP32 are administered externally to microglia cells, they could induce the production of interleukin 6 and tumor necrosis factor α and increase the phagocytosis and clearance of Aβ peptides which indicated the regulatory role of HSPs in microglia activation and neuroprotection [46]. There is a significant increase in HSP90, HSP70, and HSP27 in the activated Muller glia in proliferative vitreoretinopathy human retina, ischemia-reperfusion injury rabbit retina, and glaucomatous rat retina respectively [47, 48, 49]. The Muller HSPs could be playing role in accommodation to stress in the gliotic retina. ATP is a purine base that mediates almost all physical responses such as glucose metabolism, muscle contraction, biosynthesis, and molecular transfer within the cell [6]. However, the extracellular ATP acts as DAMPs and activates microglia and astrocytes, and induces a neuroinflammatory response. The ATP shows concentration- dependent activation of astrocytes in terms of cell proliferation, stellation, shape remodeling, and underlying actin and GFAP filament rearrangement [50].
The activated Muller glia releases ATP which indeed induces overexpression of P2X7R in retinal ganglionic cells resulting in ganglionic cell death. The intravitreal ATP injection has also the similar result of P2X7R upregulation and ganglionic cell death [8]. Cyclophilin A is a multifunctional protein that is involved in a variety of inflammatory situations. The extracellular cyclophilin A significantly enhances astroglia and microglia activation in the brain. Fragments of cyclophilin A from scrapie-infected brains are a potent stimulator of cytokine release by both microglia and astroglia in vitro and treatment with Cyclosporin A (an inhibitor of Cyclophilin A) or Cyclophilin A antibody can inhibit gliosis and cytokines production by microglia and astroglia [51]. However, the role of cyclophilin A in Muller glia has not been studied yet. A𝛽 induced gliosis and inflammation with the release of neurotoxic cytokines in Alzheimer’s diseases brain and prominently contribute to the progression of the disease [52]. The intravitreal injection of Aβ causes neurotoxicity with a significant decrease in photoreceptors and ganglionic cells and an increase in Muller cell gliosis [9]. The plasma membrane, endoplasmic reticulum, and mitochondrial DAMPs, though reported to play important role in retinal and neuronal diseases, however, their role in gliosis is not elucidated yet.
Nuclear DAMPs and Gliosis
The multiple nuclear DAMPs such as histone, HMGB-1, HMG-N1, IL-1α, IL-33, DNA, and RNA are reported to involve in neuronal and retinal diseases, however, only histones and HMGB1 are reported to play important role in gliosis [6, 7]. Histones are highly conserved, alkaline, positively charged proteins located mainly in the nucleus. The nuclear histones H2A, H2B, H3, and H4 form nucleosomes, and H1 and H5 form linkers and they play important role in regulating nuclear architecture. However, histones can also be released into the extracellular space by both damaged and activated cells, evincing significant toxic or pro-inflammatory activity both in vivo and in vitro [53]. There is a significant increase in histone proteins (H3,H4) in the gliotic proliferative vitreopathy retina as compared to the normal retina, and Muller glia could be one of the main sources of enhanced extracellular histones [47]. The histone H3 is found on the outer side of the detached retina and is associated with photoreceptor death in the rat model [54]. The overexpression of H1 in the diabetic retina has been reported to cause gliosis and promotes autophagy and neuronal cell death in the retina. Histone H1 promotes autophagy by upregulating SIRT1 and HDAC1 to maintain the deacetylation status of H4K16, leads to upregulation of ATG proteins, then promotes autophagy in the cultured retinal cell line [55]. The damaged brain also secrets histone H1 to the extracellular space which activates microglia and astrocytes and leads to neuronal cell death. [56] In Alzheimer’s disease, the non-nuclear stone H1 is found on the cell surface of neurons and activated astrocytes. However, the mechanism of the molecular mechanism of gliosis by histones is not known [57]. HMGB1, a member of the HMG protein family, which is located in the cell nucleus, has a critical function in DNA transcription and regulation. When it is released to the extracellular space, HMGB1 is known to induce inflammation by activating the NF-κB pathway by binding to TLR2, TLR4, TLR9, and the receptor for advanced glycation end products (RAGE) [6, 58]. HMGB-1 expressions and release to extracellular space are significantly enhanced in the ischemic brain resulting in neuroinflammation. The extracellular HMGB-1 plays a crucial role in microglia activation in ischemic brain as well as in vitro cell culture, whereas the treatment with HMGB-
1 antibody or HMGB shRNA significantly reduced microglia inhibition and inflammation [59]. In diabetic retinopathy, HMGB-1 plays an important role in vascular permeability by inducing microglia and astrocyte activation. The activated microglia and astrocytes secrete pro-inflammatory cytokines after HMGB-1 treatment. Further, pericyte death can be mediated by HMGB-1-induced cytotoxic activity of glial cells, whereas HMGB-1 can directly mediate the death of endothelial cells [4]. HMGB-1 treatment enhances STAT- 3 expression in Muller glia and plays a crucial role in VEGF secretion by Muller glia and endothelial cell migration, though there is no study found on the direct role of HMGB- 1 in Muller gliosis yet. Further, glycyrrhizin an HMGB-1 inhibitor attenuates the diabetes-induced upregulation of pSTAT-3 in the retina and hence inhibits HMGB1-induced VEGF secretion by Müller cells and HRMEC migration [60]. HMGB-1 is reported to significantly enhanced expression in aging astrocytes but is reduced in aging neurons. This finding is correlated with the enhanced DNA damage and neuronal cell death and enhanced proliferation of astrocytes with aging suggesting its role in neuronal aging and degeneration [61]. The circulating cell-free DNA significantly increases the traumatic brain injury, however, it could be playing important role in neurodegeneration and gliosis [62].
Conclusions
In recent years, much has been known about the role of DAMPs in neuronal diseases and both brain and retinal injury. Though the gliosis is important in neuronal injury, inflammation, and degeneration, the basic understanding of these DAMPs on gliosis is in the naïve stage. In this regard, understanding the implications of DAMP-mediated neuroinflammation and neurodegeneration could provide valuable knowledge in exploring possible diagnostic and therapeutic approaches aimed at mitigating the progress of neurodegenerative pathologies.
Acknowledgments
The author acknowledges support and inspiration from the Ocular Immunology and Angiogenesis Laboratory, ARVO EyeFind grant, and Professional Development award at Medical College of Wisconsin, USA. However, no funding was received for this manuscript from any funding agencies.
Disclosure
The author reports no conflicts of interest in this work
Data Availability
The authors shall make available the data underlying this study on request.
References
-
Buffo A, Rite I, Tripathi P, Lepier A, Colak D, et al. (2008) Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci USA 105(9): 3581-3586.
-
Kang S, Larbi D, Andrade M, Reardon S, Reh TA, et al. (2021) A Comparative Analysis of Reactive Müller Glia Gene Expression After Light Damage and microRNA- Depleted Müller Glia Focus on microRNAs. Front Cell Dev Biol 8: 620459.
-
Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81(2): 229-248.
-
Santos ARC, Dvoriantchikova G, Li Y, Mohammad G, Abu El-Asrar AM, et al. (2014) Cellular mechanisms of high mobility group 1 (HMGB-1) protein action in the diabetic retinopathy. PloS one 9(1): e87574.
-
Pannicke T, Wagner L, Reichenbach A, Grosche A (2018) Electrophysiological characterization of Müller cells from the ischemic retina of mice deficient in the leukemia inhibitory factor. Neurosci Lett 670: 69-74.
-
Thundyil J, Lim KL (2015) DAMPs and neurodegeneration. Ageing Res Rev 24(Pt A): 17-28.
-
Mahaling B, Low SWY, Beck M, Kumar D, Ahmed S, et al. (2022) Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders. Int J Mol Sci 23(5): 2591.
-
Xue Y, Xie Y, Xue B, Hu N, Zhang G, et al. (2016) Activated Müller Cells Involved in ATP-Induced Upregulation of P2X(7) Receptor Expression and Retinal Ganglion Cell Death. Biomed Res Int 16: 9020715.
-
Walsh DT, Montero RM, Bresciani LG, Jen AYT, Leclercq PD, et al. (2002) Amyloid-Beta Peptide Is Toxic to Neurons In Vivo via Indirect Mechanisms. Neurobiol Dis 10(1): 20-27.
-
Stephenson EL, Mishra MK, Moussienko D, Laflamme N, Rivest S, et al. (2018) Chondroitin sulfate proteoglycans as novel drivers of leucocyte infiltration in multiple sclerosis. Brain 141(4): 1094-1110.
-
Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, et al. (2010) Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 58(3): 300-314.
-
Logan A, Baird A, Berry M (1999) Decorin attenuates gliotic scar formation in the rat cerebral hemisphere. Exp Neurol 159(2): 504-510.
-
Botfield H, Gonzalez AM, Abdullah O, Skjolding AD, Berry M, et al. (2013) Decorin prevents the development of juvenile communicating hydrocephalus. Brain 136(Pt 9): 2842-2858.\
-
Schneider M, Dillinger AE, Ohlmann A, Iozzo RV, Fuchshofer R (2021) Decorin-An Antagonist of TGF-β in Astrocytes of the Optic Nerve. Int J Mol Sci 22(14): 7660.
-
Lam V, Takechi R, Pallebage-Gamarallage MM, Galloway S, Mamo JC (2011) Colocalisation of plasma derived apo B lipoproteins with cerebral proteoglycans in a transgenic- amyloid model of Alzheimer’s disease. Neurosci Lett 492(3): 160-164.\
-
Stichel CC, Kappler J, Junghans U, Koops A, Kresse H, et al. (1995) Differential expression of the small chondroitin/ dermatan sulfate proteoglycans decorin and biglycan after injury of the adult rat brain. Brain Res 704(2): 263- 274.
-
McKeon RJ, Jurynec MJ, Buck CR (1999) The Chondroitin Sulfate Proteoglycans Neurocan and Phosphacan Are Expressed by Reactive Astrocytes in the Chronic CNS Glial Scar. J Neurosci 19(24): 10778-10788.
-
Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182(2): 399- 411.
-
Matsuyama A, Kalargyrou AA, Smith AJ, Ali RR, Pearson RA (2022) A comprehensive atlas of Aggrecan, Versican, Neurocan and Phosphacan expression across time in wildtype retina and in retinal degeneration. Scientific Reports 12(1): 7282.
-
Chan CCM, Roberts CR, Steeves JD, Tetzlaff W (2008) Aggrecan components differentially modulate nerve growth factor–responsive and neurotrophin-3- responsive dorsal root ganglion neurite growth. J Neurosci Res 86(3): 581-592.
-
Eastlake K, Banerjee P, Angbohang A, Charteris D, Khaw P, et al. (2016) Müller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 64(4): 495-506.
-
Crapser JD, Spangenberg EE, Barahona RA, Arreola MA, Hohsfield LA, et al. (2020) Microglia facilitates loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58: 102919.
-
Struve J, Maher PC, Li YQ, Kinney S, Fehlings MG, et al. (2005) Disruption of the hyaluronan-based extracellular matrix in spinal cord promotes astrocyte proliferation. Glia 52(1): 16-24.
-
Chistyakov DV, Astakhova AA, Azbukina NV, Goriainov SV, Chistyakov VV, et al. (2019) High and Low Molecular Weight Hyaluronic Acid Differentially Influences Oxylipins Synthesis in Course of Neuroinflammation. Int J Mol Sci 20(16): 3894.
-
Liuzzo JP, Petanceska SS, Moscatelli D, Devi LA (1999) Inflammatory Mediators Regulate Cathepsin S in Macrophages and Microglia: A Role in Attenuating Heparan Sulfate Interactions. Mol Med 5(5): 320-333.
-
Park PJ, Shukla D (2013) Role of heparan sulfate in ocular diseases. Exp Eye Research 110: 1-9.
-
Properzi F, Lin R, Kwok J, Naidu M, van Kuppevelt TH, et al. (2008) Heparan sulphate proteoglycans in glia and in the normal and injured CNS: expression of sulphotransferases and changes in sulphation. Eur J Neurosci 27(3): 593-604.
-
Sobel RA, Mitchell ME (1989) Fibronectin in multiple sclerosis lesions. Am J Pathol 135(1): 161-168.
-
Price J, Hynes RO (1985) Astrocytes in culture synthesize and secrete a variant form of fibronectin. The Journal of neuroscience: the official journal of the Society for Neuroscience 5(8): 2205-2211.
-
Orr MB, Gensel JC (2018) Spinal Cord Injury Scarring and Inflammation: Therapies Targeting Glial and Inflammatory Responses. Neurotherapeutics 15(3): 541-553.
-
Kim H, Ahn M, Choi S, Kim M, Sim KB, et al. (2013) Potential role of fibronectin in microglia/macrophage activation following cryoinjury in the rat brain: an immunohistochemical study. Brain Res 1502: 11-19.
-
Kanda A, Noda K, Hirose I, Ishida S (2019) TGF-β-SNAIL axis induces Müller glial-mesenchymal transition in the pathogenesis of idiopathic epiretinal membrane. Scientific Reports 9(1): 673.
-
Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, et al. (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF- beta after vascular damage. J Neurosci 30(17): 5843- 5854.
-
Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, et al. (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nature Communications 3(1): 1227.
-
Stichel CC, Müller HW (1994) Relationship between injury-induced astrogliosis, laminin expression and axonal sprouting in the adult rat brain. J Neurocytol 23(10): 615-630.
-
Tam WY, Au NPB, Ma CHE (2016) The association between laminin and microglial morphology in vitro. Scientific Reports 6: 28580-28580.
-
Niquet J, Jorquera I, Faissner A, Ben-Ari Y, Represa A (1995) Gliosis and axonal sprouting in the hippocampus of epileptic rats are associated with an increase of tenascin-C immunoreactivity. J Neurocytol 24(8): 611- 624.
-
Wiemann S, Reinhard J, Reinehr S, Cibir Z, Joachim SC, et al. (2020) Loss of the Extracellular Matrix Molecule Tenascin-C Leads to Absence of Reactive Gliosis and Promotes Anti-inflammatory Cytokine Expression in an Autoimmune Glaucoma Mouse Model. Frontiers in Immunology 11: 1-21.
-
Haage V, Elmadany N, Roll L, Faissner A, Gutmann DH, et al. (2019) Tenascin C regulates multiple microglial functions involving TLR4 signaling and HDAC1. Brain Behavior Immunity 81: 470-483.
-
Sugiura S, Lahav R, Han J, Kou SY, Banner LR, et al. (2000) Leukaemia inhibitory factor is required for normal inflammatory responses to injury in the peripheral and central nervous systems in vivo and is chemotactic for macrophages in vitro. Eur J Neurosci 12(2): 457-66.
-
Renkawek K, Stege GJJ, Bosman GJCGM (1999) Dementia, gliosis and expression of the small heat shock proteins hsp27 and αB-crystallin in Parkinson’s disease. Neuroreport 10(11): 2237-2236.
-
Kerr BJ, Patterson PH (2004) Potent pro-inflammatory actions of leukemia inhibitory factor in the spinal cord of the adult mouse. Exp Neurol 188(2): 391-407.
-
Cirillo C, Capoccia E, Iuvone T, Cuomo R, Sarnelli G, et al. (2015) S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. Biomed Res Int 508342.
-
Casola C, Schiwek JE, Reinehr S, Kuehn S, Grus FH, et al. (2015) S100 alone has the same destructive effect on retinal ganglion cells as in combination with HSP 27 in an autoimmune glaucoma model. J Mol Neurosci 56(1): 228-236.
-
Shao X, Lu W, Gao F, Li D, Hu J, et al. (2016) Uric Acid Induces Cognitive Dysfunction through Hippocampal Inflammation in Rodents and Humans. The Journal of neuroscience: the official J Neurosci 36(43): 10990- 11005.
-
Kakimura JI, Kitamura Y, Takata K, Umeki M, Suzuki S, et al. (2002) Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J 16(6): 601-603.
-
Eastlake K, Heywood WE, Banerjee P, Bliss E, Mills K, et al. (2018) Comparative proteomic analysis of normal and gliotic PVR retina and contribution of Muller glia to this profile. Exp Eye Res 177: 197-207.
-
Kalesnykas G, Niittykoski M, Rantala J, Miettinen R, Salminen A, et al. (2007) The expression of heat shock protein 27 in retinal ganglion and glial cells in a rat glaucoma model. Neuroscience 150(3): 692-704.
-
Gohdo T, Ueda H, Ohno S, Iijima H, Tsukahara S (2001) Heat shock protein 70 expression increased in rabbit Müller cells in the ischemia-reperfusion model. Ophthalmic Res 33(5): 298-302.
-
Adzic M, Stevanovic I, Josipovic N, Laketa D, Lavrnja I, et al. (2017) Extracellular ATP induces graded reactive response of astrocytes and strengthens their antioxidative defense in vitro. J Neurosci Res 95(4): 1053-1066.
-
Tanveer DT, Carroll JA, Moore RA, Striebel JF, Chesebro B (2012) Role of cyclophilin A from brains of prion-infected mice in stimulation of cytokine release by microglia and astroglia in vitro. J Biol Chem 287(7): 4628-4639.
-
Craft JM, Watterson DM, Van Eldik LJ (2006) Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 53(5): 484-490.
-
Chen R, Kang R, Fan XG, Tang D (2014) Release and activity of histone in diseases. Cell Death Dis 5(8): e1370-e1370.
-
Kawano H, Ito T, Yamada S, Hashiguchi T, Maruyama I, et al. (2014) Toxic effects of extracellular histones and their neutralization by vitreous in retinal detachment. Lab Invest 94(5): 569-585.
-
Wang W, Wang Q, Wan D, Sun Y, Wang L, et al. (2017) Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy 13(5): 941-954.
-
Gilthorpe JD, Oozeer F, Nash J, Calvo M, Bennett DL, et al. (2013) Extracellular histone H1 is neurotoxic and drives a pro-inflammatory response in microglia. F1000Research 2: 1-14
-
Bolton SJ, Carneiro MR, Betmouni S, Perry VH (1999) Non-nuclear histone H1 is upregulated in neurones and astrocytes in prion and Alzheimer’s diseases but not in acute neurodegeneration. Neuropathol Appl Neurobiol 25(5): 425-432.
-
Roh JS, Sohn DH (2018) Damage-associated molecular patterns in inflammatory diseases. Immune netw 18(4): e27
-
Kim JB, Choi JS, Yu YM, Nam K, Piao CS, et al. (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26(24): 6413-6421.
-
Mohammad G, Jomar D, Siddiquei MM, Alam K, Abu El Asrar AM (2017) High-Mobility Group Box-1 Protein Mediates the Regulation of Signal Transducer and Activator of Transcription-3 in the Diabetic Retina and in Human Retinal Muller Cells. Ophthalmic Res 57(3): 150-160.
-
Enokido Y, Yoshitake A, Ito H, Okazawa H (2008) Age- dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem Biophys Res Commun 376(1): 128-133.
-
Rodrigues Filho EM, Simon D, Ikuta N, Klovan C, Dannebrock FA, et al. (2014) Elevated cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J Neurotrauma 31(19): 1639-1646.
- Are the Vaccines the Only Solution to Prevent the COVID-19 Pandemic? Part Two
- Clinical Characteristics of Women in this New Global Immunodeficiency
- Cell Dynamics in HIV Pathogenesis: Insights and Implications
- Determination of the CDR (CDR1, CDR2) « Complementary- Determining Region Invertebrate Primitive Antibody from Sea Star »
- Prioritizing Care for High-Risk COVID-19 Patients in the EU: 10 Civic Recommendations to the Institutions
- Comprehensive Insights into ModRNA Vaccines: Persistent PP-Spike Recombinant Protein, Hyperimmune/Inflammatory Reactions, Thrombotic Vasculopathy, Chronic Organ Complications and Excess Deaths