Advancements in Neurological Gene Therapy
Neurological disorders can be extremely harmful and affect a large portion of the population, with the elderly being particularly susceptible. Gene mutations that cause aberrant development of the nervous system, neurodegeneration, or reduced neuronal function are frequently the origin of these disorders. Genetic and epigenetic alterations induced by environmental stressors, trauma, illness-related events, or inflammatory processes are additional causes of neurological disorders. The use of conventional medicine and surgery to treat or cure certain illnesses has not shown to be successful, as there are either no suitable medications available or not enough of them to stop the disease's progression. Gene therapy is becoming a more potent method that may be able to treat and even cure some of the most prevalent nervous system illnesses. Advances in the design of gene vectors, therapeutic gene selection, and delivery techniques, along with a better knowledge of the underlying disease mechanisms—particularly those involving sensory neurons—have made gene therapy for neurological illnesses feasible. Developments in the field restored our hope for neurologists, ophthalmologists, and neurosurgeons to utilise gene therapy as a therapeutic option. In this Review, we address the prospects for further advancements in gene therapy development as well as the prospective gene therapy procedures that may be used to treat patients with neurological illnesses.
Introduction
The brain is an organ where many of the most common illness processes originate, for which the aetiology is still unknown. The nervous system is a complicated and challenging organ system to investigate. These illnesses can affect metabolism, brain development, and function on a local or global scale [1]. They cover a wide range of pathological disorders. Due to the complexity and incomplete understanding of the underlying pathophysiology of these illnesses, medications and neurosurgical techniques have generally not shown to be useful in their treatment. Furthermore, because it prevents therapeutic medicines from being widely delivered to the central nervous system (CNS), the blood–brain barrier (BBB) restricts the use of systemic therapy.
An alternative to conventional pharmacological treatments could be genetic interventions that provide gene products that induce the restoration of destroyed cells and/or permanently restore function [2]. Gene therapy refers to such methods, which employ DNA or RNA as the pharmacological agent. Despite a difficult beginning, gene therapy has developed and is now regarded as a respectable and promising option for treating a variety of nervous system illnesses [3]. The development of advanced delivery vehicles that have been tested in animal models of human disease is largely responsible for advancements in gene transfer techniques [4]. Early clinical trials have been conducted based on a multitude of preclinical investigations to evaluate the safety and, in certain situations, effectiveness of gene therapy. Several positive outcomes imply that this strategy may soon be implemented in the clinic.
Some of the most promising new gene therapy techniques for the treatment of diverse nervous system illnesses are discussed in this review. We start by outlining the most popular gene delivery methods and how vector biology and design complement each other. In the PNS, we discuss advancements in the treatment of polyneuropathy, neuropathic pain, and retinal degeneration; in the CNS, we concentrate on glioblastoma, Parkinson disease (PD), lysosomal storage disorders, and epilepsy [5]. While other conditions have seen progress in the field of gene therapy, the diseases mentioned above serve as a good example of how the nervous system’s gene therapy has developed.
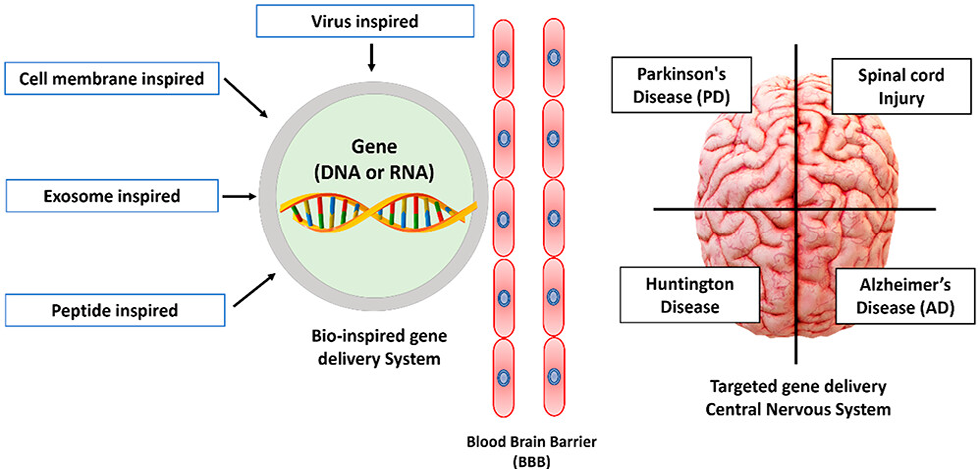
In our view, gene therapy may be able to prevent neurological illnesses from developing, delay their progression, or even restore normal function [6]. We anticipate that some of these gene therapy techniques will be made available to patients in the future. Figure 1 shows the Current Challenges Involving Cns-Targeted Gene Delivery.
Complexity of the Brain Structure
Despite the complex and diverse structure of the brain, neurons continue to be the most studied type of central nervous system (CNS) cell. Recent developments in biomedicine, however, are revealing the complex relationships between the pathophysiology of numerous neurological illnesses and the functions of different cell subpopulations inside the brain [7]. The resident immune cells of the central nervous system (CNS), known as microglia, comprise approximately 10- 15% of all brain cells and are responsible for regular brain surveillance. In the central nervous system, oligodendrocytes myelinate axons and electrically insulate them, enabling saltatory conduction, which speeds up nerve conduction. Fig. 2 represents the Complexity of the Brain Structure.
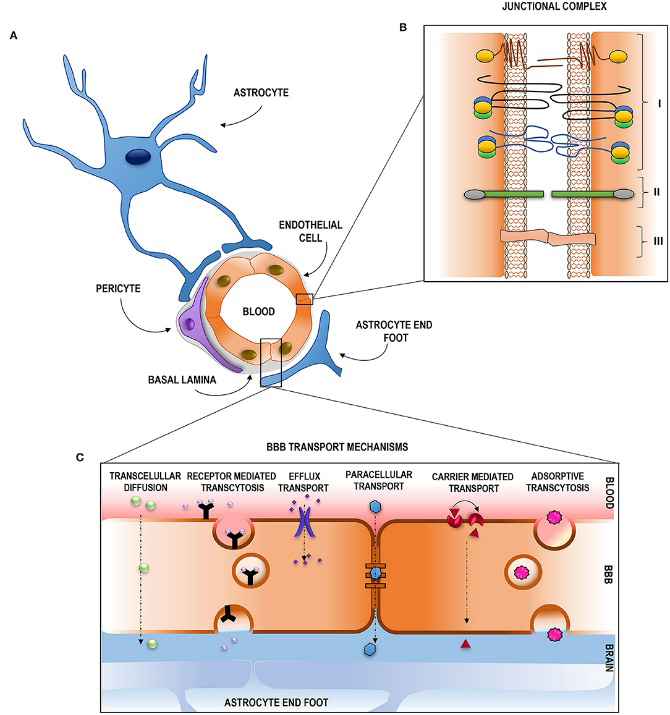
The primary glial cells and the most prevalent kind of cells in the brain are called astrocytes. These glial cells are essential to the structural component of the blood–brain barrier (BBB) that surrounds the tight junctions. Due to the distinct aetiology of neurological illnesses, delivery methods for gene therapy that can target particular cell subpopulations are needed. Several methods, including parenchymal injections, have been used to administer these targeted gene delivery. The fundamental issue that needs to be resolved is determining the brain’s global gene expression [8].
Low Permeability of the Blood–Brain Barrier
Treatment for neurological illnesses is challenging because brain areas shielded by physical barriers like the blood-brain barrier are difficult to reach. The BBB serves as a barrier to keep the central nervous system (CNS) stable and prevent toxins from entering into the brain. The primary cellular components of this barrier are pericytes, perivascular glial processes, and a monoendothelial layer of cells joined by tight junctions; the basal lamina envelops these components. Pinocytotic vesicles and the comparatively small number of fenestrations on the endothelial cells which compose up the BBB further limit the passage of chemicals across it and into the central nervous system [9].
Small enough or fat soluble molecules can passively traverse the blood-brain barrier. The BBB cells’ expressed receptors, transporters, or carriers may actively absorb them and allow them to cross. The brain’s highly arborized blood artery network allows for the parenchyma to be globally perfused because every brain cell is located within 20 mm of a blood capillary. Intravascular gene delivery devices might potentially penetrate the entire brain if the blood-brain barrier could be crossed, given the brain’s large vascular anatomy. Targeting brain cells is physically difficult due to the BBB’s poor permeability [10]. Therefore, a small size, high lipophilicity, and minimal plasma protein binding would be required for the excellent therapeutic agent. Other routes of administration have been used to get around the BBB. Figure 3 shows Permeability of the Blood–Brain Barrier.
In addition, there are extracellular and intracellular barriers that prevent the uptake of genetic material. Among the extracellular barriers is the ability to evade the host immunological reaction. Segmental escape and vesicle destiny are examples of intracellular barriers. Only ~2% of nucleic acid escapes the endocytic vesicle, despite the fact that most cells will internalise vectors. The endosome, a hostile environment for nucleic acids, is where nanoparticles will be localised. Nucleic acids should reach the compartment of interest after endosomal escape and cytoplasmic release, but the mobility of bigger molecules is very constrained [11].
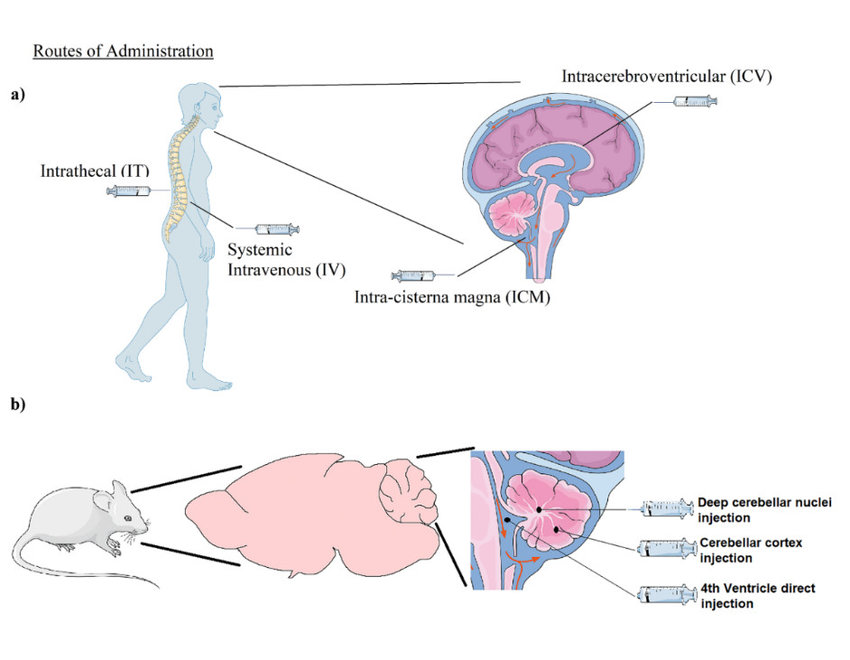
Routes of Administration
It is challenging to determine the pharmacokinetic and pharmacodynamic characteristics for gene therapy since the dose and frequency depend on the delivery mechanism used in addition to the gene being introduced. Gene therapy administration methods have been investigated, including intrathecal, intramuscular, intranasal, and intracarotid. The most effective administration methods at this time have been demonstrated to be intravenous and intracerebral.
Current research is focused on developing systems via the intravenous route (Figure 4) because it is the most convenient for administration. Although administering medication intracerebrally provides a targeted therapeutic approach, there have been reported cases of brain diffusion into unwanted sites [12]. The shift from in vitro to in vivo investigations and from mice to nonhuman primates presents another challenge in establishing the pharmacokinetic and pharmacodynamic profile for gene therapy. Dosing paradigms and delivery system designs are known to differ significantly amongst models [13].
Modes of Gene Delivery
Gene therapy can modify genes in a variety of ways, including inserting a new gene into the body to aid in the fight against a disease, inactivating or “knocking out” a mutated gene that functions abnormally, or replacing a mutated gene that causes disease with a healthy copy of the gene [14]. Gene augmentation, silencing, and editing as gene delivery mechanisms will be covered in the section that follows. Figure 5 represents the Modes of Gene therapy.
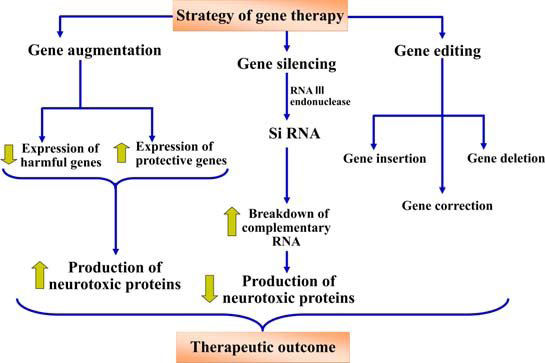
Gene Augmentation
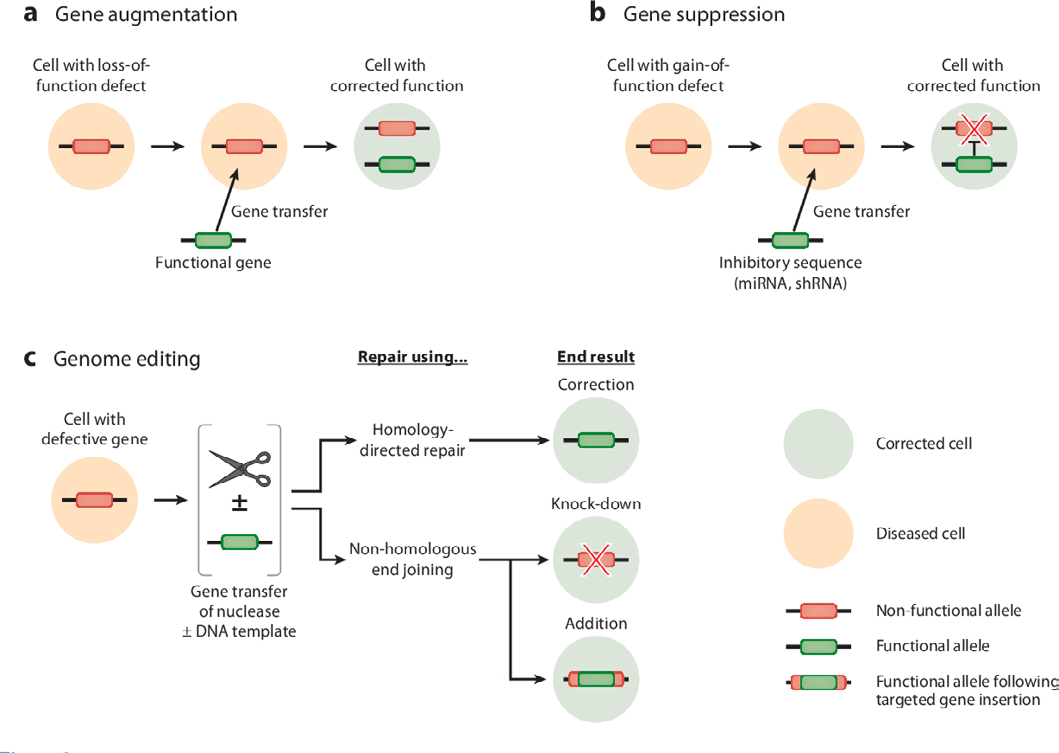
Increased expression of a target gene is achieved through gene augmentation. When particular genes are down-regulated or a missense mutation is inserted, gene augmentation (Figure 6) is used. This results in a non- functional protein being translated. Studies implementing the gene augmentation modality to enhance neuroprotective and neurorestorative genes, specifically in Alzheimer’s, Parkinson’s, and Huntington conditions, have been described. This modality can be utilised to increase protein levels [15].
As a result of AD, astrocytes become persistently “activated” cells with morphological and biochemical alterations that appear to impair their protective qualities and perhaps exacerbate neuroinflammatory processes. A study that targeted hippocampus astrocytes using adeno- associated virus (AAV) vectors that included the astrocyte- specific Gfa2 promoter. Increased expression of VIVIT, a peptide that disrupts the immune/inflammatory calcineurin/ NFAT pathway, was seen in APP/PS1 mice. Following several months of Gfa2-VIVIT treatment, APP/PS1 animals showed decreased glial activation, decreased amyloid levels— which are key in defining the pathophysiology of AD—and enhanced cognitive function. Neurotrophic levels, including brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), and nerve growth factor (NGF), have been shown to be downregulated in a number of neurological diseases; replacing these proteins through gene augmentation has been investigated as a potential treatment. Dopamine-containing neurons (DA) in the substantia nigra (SN) that project to the striatum gradually disappear, which is a hallmark of Parkinson’s disease [16]. In a rat model of Parkinson’s disease, studies have shown that GDNF can preserve DA neurons when administered via a viral vector. Huntington’s disease is associated A study that used transgenic mice overexpressing BDNF under the glial fibrillary acidic protein (GFAP) promoter investigated the intrastriatal delivery of quionolinate. When quionolinate was administered to pGFAP-BDNF mice instead of wild- type mice, it was found to increase the quantity of reactive astrocytes and improve BDNF release [17]. When compared to animals who had wild-type astrocytes implanted into them, these mice’s conduct improved gradually. According to these results, BDNF may be used as a treatment for Huntington’s disease with the neuroprotective protein BDNF [18].
Research has demonstrated that augmenting the amounts of an inhibitor can be employed therapeutically, in addition to promoting the expression of genes known to be neuroprotective. It has been demonstrated that overexpressing 7ND, a dominant negative counterpart of CCL2m, reduces the proinflammatory effects in amyloid precursor protein-presenilin-1 in mouse models of AD. After six months, 7ND expression in bigenic mice for APP/ presenilin-1 (APP/PS1) decreased beta-amyloidosis and astro-microgliosis while improving spatial learning [19].
Gene silencing may decrease the concentrations of neurotoxic proteins that exacerbate neuroinflammation as well as can lessen the synthesis of proteins that act inappropriately. Gene silencing can be achieved by post-transcriptional control of a gene using the RNA (Figure 7) interference approach, which is beneficial for neuropathological conditions caused by nonsense or repetitive mutations. Small interfering RNAs (siRNAs) are produced in vivo by the RNase III endonuclease Dicer in a multistep process known as RNAi [20].
The complementary RNA is degraded by the ensuing 21–23 nucleotide siRNAs. The primary mechanism of siRNA- directed mRNA degradation is the formation of the siRNA- induced silencing complex (RISC). A study examined the effectiveness of e-PAM-IR-mediated siRNA delivery in rat brains and primary cortical cultures.
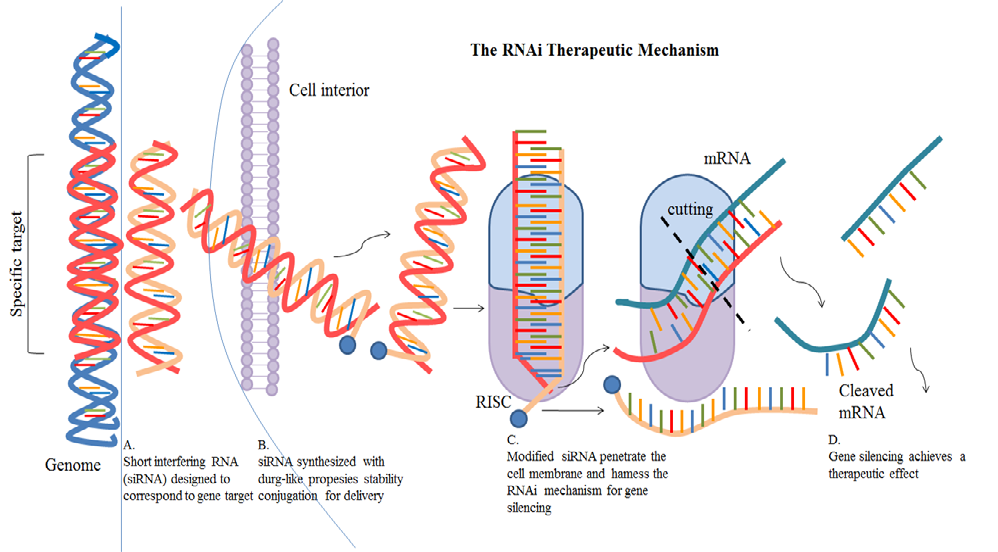
After transfecting High mobility group box-1 (HMGB1, a new cytokine-like molecule) with siRNA into primary cortical cultures treated with NMDA or H2O2, the potential utility of e-PAM-R was established by gene knockdown. Neuronal cell death was considerably inhibited in both cases of H2O2 - or NMDA-induced HMGB1 levels, which were successfully lowered by HMGB1 siRNA administration. Lentiviral- mediated suppression of GFAP and vimentin in astrocytes is another method of using RNA interference (RNAi) for gene silencing. The glial scar that prevents axonal regeneration following CNS injury is formed by the overexpression of two intermediate filament proteins by reactive astrocytes. Cortical neurons in this work exhibited high survival rates and improved neurite development when cultivated; knockdown of GFAP and vimentin using RNAi lowered astrocyte responsiveness and consequent glial scarring. Brain oedema is related to astrocytes’ production of aquaporin 4 (AQP4) [16]. A therapeutic decrease in cerebral water build- up has been proposed as a result of the elimination of these porins. In astrocyte primary cultures, AQP4 expression was suppressed by using siRNA. According to this study, AQP4 appears to be in charge of the rapid water transfer in astrocytes that have been cultivated. Additionally, the cDNA microarray analysis demonstrated that AQP4 gene silencing impacted other gene expression patterns, particularly those related to ischemia, like GLUT1 and hexokinase. According to this study, ischemia-related protein expression may be decreased by blocking astrocyte-AQP4 water channels [21].
Editing
The most recent gene therapy modality to be identified is gene editing. Engineered nucleases made of non-specific DNA cleavage module linked to sequence-specific DNA binding domains are employed for carrying it out. By causing exact double strand breaks in DNA, these nucleases efficiently and precisely alter DNA and activate cellular DNA repair systems. The transcription activator like effector nucleases (TALENs), zinc finger nucleases (ZFNs), and clustered regulatory interspaced short palindromic repeats (CRISPR)/ Cas 9 are examples of these endonucleases. Double-stranded breaks caused by ZFNs and TALENs at particular genomic sites promote error-prone non-homologous end-joining or homology-directed repair [22]. Huntington’s disease is caused by a mutation in the htt gene that results in an increase in CAG repeats at the 50 end of the gene. Since it is unknown how much htt gene is needed to support normal neuronal activity, knocking down the gene would not be therapeutically beneficial. Since the severity of the condition is correlated with the amount of CAG repeats, gene editing (Figure 8) is the best method for eliminating overexpressed CAG repeats. Mutant proteins can be eliminated or mutated genes can be corrected in vivo using TALENS nucleases and zinc finer [23]. ZFN overlapping CAG repeats were given to R6/2 mice to suppress aggressive behaviours linked to the advancement of the disease and to downregulate the Human Huntington mRNA. Furthermore, CRISPR using the Cas9/ guide RNA (gRNA) method was employed in a study to remove the integrated HIV-1 genome in HIV-infected patients [24].
Cas9/gRNA identified and altered highly precise targets within the HIV-1 LTR U3 region, thereby inactivating viral gene expression and preventing latently infected T-cells and microglia from replicating. In HIV-associated neurodegenerative illness, the brain’s astrocytes and microglia develop into viral reservoirs that persistently release viral proteins that impair neurotoxicity and neurocognitive function [25].
Vectors for Gene Therapy
Gene delivery must be efficient for gene therapy to be successful. The design of therapeutic gene delivery vectors has changed significantly over the last 20 years in order to satisfy the complex demands of transgene delivery to the host. Due to their low cost and enormous cargo delivery capacity, nonviral gene delivery vehicles such as liposomes, nanoparticles, and bare DNA or RNA have been the subject of much research and development [26]. However, therapeutic gene expression using these vectors is usually modest and short-lived. The highly developed methods that wild-type viruses employ to introduce their genetic material into host cells and change the functions of those cells are absent from all of these nonviral vectors. Figure 9 shows the Viral and non-viral vectors.
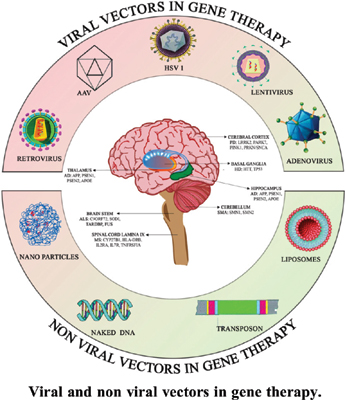
Vectors for Viruses
Utilising the biology of the virus for transgenic expression rather than viral replication following host transduction is essential for the development of an efficient viral vector. The achievement of this objective has proven to be challenging, and appropriate vectors continue to be developed, differing in terms of cellular specificity, safety concerns, and the degree and duration of transgenic expression. For instance, vectors made from AAVs allow for long-term gene expression and are safe and non-pathogenic. However, these vectors are easily destroyed by humoral immune responses in individuals who have already been exposed to the virus, have a limited capacity to transgene, and can be challenging to target to the right area. They also require a high dose to effectively express a gene [27]. To prevent therapeutic gene loss in proliferating cells, alternative vectors like lentiviruses and retroviruses can introduce new genetic material into the host cell chromosome. However, using these viral vectors may have drawbacks due to oncogenesis caused by the chromosomal insertion of the vector DNA. Adenovirus and HSV-derived vectors are among the vectors with a high transgenic capacity because they can be used for efficient gene targeting and long-term transgene expression. However, because to “leaky” viral gene expression and the body’s sensitivity to the vector coat, these vectors can be toxic and induce inflammation [28].
Complete vector genome silencing, which may alter transgene expression, is necessary to prevent these negative effects. Immune-mediated inflammatory processes that restrict vector delivery, gene expression, and the possibility of redosing may be triggered by innate immune responses directed against any of these vectors in conjunction with humoral immune responses. Even with these drawbacks, a lot of these vectors have shown to be quite successful gene delivery vehicles when handled carefully and in a way that makes the most of their innate biological advantages [29].
Vectors for Viruses: Delivery Systems
Effective and secure vectors for transfecting human cells with the desired therapeutic genetic material are essential to the effectiveness of gene therapy. Significant developments in gene therapy have concentrated on enhancing safety profiles and vector designs for targeted gene delivery and practical uses of results. Among these technological advancements are plasmid transfection, nanoparticles, polymer-mediated gene delivery, tailored microRNA, highly precise viral vector designs, and in vivo CRISPR-based medicines [30].
Viruses are easily adaptable vectors for gene delivery because their genetic material is natively adapted to infect human cells. The process of designing viruses to transfer genetic sequences to human cells efficiently and without causing harm is known as viral transduction. Reliable transmission and excellent specificity for a particular cell type are the primary benefits of viral vectors [31]. Limitations include the therapeutic genetic material’s small carrying capacity, invasive administration, and the possibility of unfavourable immune reactions or downstream genetic effects. Many different viruses, each with specific advantages, limitations, and uses, have been used as vectors. Adenovirus and adeno-associated virus (AAV) are examples of current viral vector. Herpes simplex virus (HSV), lentivirus, and other retroviruses [32].
Adenovirus
Adenoviruses are viruses that include naked double- stranded DNA. Due of ADV’s high virulence and possible CNS damage, however, its usage has been restricted [33].
Virulence was eradicated with the development of helper-dependent ADV (HD-ADV), which eliminated all viral genes. With a carrying capacity of up to 30 kB, the necessary helper virus transports the data required for reproduction. Following transfection, the helper virus cannot be totally eradicated, though. Recently, human pluripotent stem cells’ DNA has been effectively modified using HDADV, providing the way for gene therapy-based stem cell therapy [34].
Lentivirus
A lentivirus is a type of enclosed single-stranded RNA retrovirus that can express several effector molecules. Similar to that, it displays years of genetic expression by integrating into the host genome, but it can carry a somewhat bigger 8–9 kB of therapeutic genetic material [35].
Integration could be useful for transfecting cells that are dividing, but if inserted into unwanted regions of the genome, it could lead to insertional mutagenesis. By decreasing insertional mutations and insertion-caused genomic instability, novel integrase-deficient lentiviruses (IDLV) enhance the safety profile and inhibit integration. According to recent research, lentiviruses can be used to raise serum levels of amyloid precursor protein-alpha and stop behavioural and memory deficits in a mouse model of Alzheimer’s disease [36].
Adeno-Associated Virus
The most widely utilised viral vector at the moment is the enclosed single-stranded DNA virus known as adeno-associated virus (AAV). This is because of its many advantageous properties. Because AAV is non-pathogenic and unable to multiply on its own, it cannot result in significant unfavourable immune responses or superinfections. By designing the virus to have almost minimal viral genetic material, focused transcription of the desired gene can be achieved without the production of viral proteins [37]. After the genetic material is transferred, AAV does not integrate into the host genome and stays extra-chromosomal or episomal. After gene delivery, this characteristic inhibits unexpected insertional mutagenesis. However, AAV persists to show years of sustained gene expression in the human brain. Accurate cell targeting and CNS dissemination is made possible by a variety of AAV serotypes, or coating proteins. Because different capsid designs confer different tissue tropisms and distribution, AAV can be used, for example, to silence superoxide dismutase-1 (SOD1) to treat ALS, replace survival motor neuron protein (SMN) to treat spinal muscular atrophy (SMA), or silence Huntington (HTT) to treat Huntington’s disease. AAV’s primary drawback is its microscopic size, which promotes perfect tissue dispersion but restricts the size of therapeutic genetic material to less than 5 kB [38].
Herpes Simplex Virus
A double-stranded DNA virus enclosed in an envelope, the herpes simplex virus can carry the most genetic material— up to 40 kB—but its tissue penetration and bio distribution are additionally restricted by this capability [39]. HSV’s high affinity for CNS neurons, particularly sensory neurons, and its capacity to persist as a stable nuclear episome after infection are its primary benefits. Although HSV can cause a severe immunological reaction, studies have shown that eliminating the viral genetic material (Figure 10) significantly decreases cytotoxicity and stabilises the virus when it is used to treat glioblastoma in rodents in vivo.
Non-Viral Vector Delivery Systems
Although viral vectors have demonstrated considerable potential as neuronal gene carriers, their high cost, non- specific targeting, toxicity, and immunogenicity hindered their widespread application [40]. Alternative strategies have been developed to get around these restrictions, such as the use of cationic polymers, cationic lipids, tailored polypeptides, nanoparticles, and naked DNA as non-viral vector delivery modalities. It has been demonstrated that these non-viral (Figure 11) vectors are far less expensive to develop, have a better safety profile, and can target certain populations of brain cells. Significantly lower transfection efficiency than viral vectors are the drawback of non-viral vectors [41].
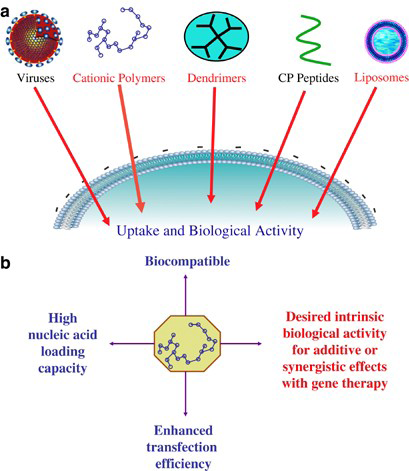
Cationic Polymers
Nucleic acids are effectively condensed into nanoparticles known as “polyplexes” by cationic polymers. It has been demonstrated that adding cationic polymers to negatively charged particles, like gold nanoparticles thiolated with siRNA, greatly increased the siRNA’s cytoplasmic transport. Gene silencing in this study was not mediated by gold-siRNA nanoparticles alone, but silencing grew to over 90% when the nanoparticles were coated with a cationic polymer [42]. Cell targeting molecules connected to the functional group of cationic polymers are hypothesised to enable the condensing of the transported DNA or RNA through electrostatic interactions and transfer of the material via ligands.
Cationic Lipids
It has been demonstrated that cationic lipids are the most effective non-viral vector because they can transport bigger nucleic acids. The different commercially available cationic lipid formulations are typically made up of a neutral lipid such as cholesterol and dioleoylphosphatidylethanolamine (DOPE), along with a cationic lipid such as 1,2-dioleoyl- 3-trimethylammonium propane (DOTAP), N-methyl-4- (dioleyl) methylpyridinium (SAINT-2), 3b-[N-(N0,N0- dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol), or GS1. Since then, new formulations have raised the transfection rate to 20–25% into primary neurons. Initially, cationic lipid for gene therapy delivery for neurological illnesses produced a transfection rate of 1-3 into primary neural cells. Because of their tendency for association in biological fluids, cationic lipids present a difficulty [43].
Engineered Polypeptides
Targeting particular cell subpopulations, endosomal escape, and nucleus entry into a single recombinant protein are all possible with engineered polypeptides. The tetanus toxin fragment C is the basis for the engineered polypeptides that are designed to use recombinant fusion proteins for neuronal targeting. Additional domains are included for endosomal escape and DNA condensation, which are mediated by the translocation domain of the diphtheria toxin and the GAL4 transcription factor DNA-binding domain, respectively [44].
Using a targeted peptide generated from NGF was reported in another investigation. Using chimeric complexes made of either NGF loop 4 or an NGF hairpin motif comprising loops 1 and 2 fused to a DNA-binding domain, neuron- specific transfection was accomplished in the previously stated investigations [45].
Nanoparticles
The cost-effective production process and safety profile of nanoparticles render them a promising gene therapy delivery strategy. By binding to DNA and mediating reporter gene expression in neurons and glia through intravenous injections, iron oxide nanoparticles coated with PLL demonstrated the transport of plasmid DNA into cultured neurons. An additional investigation revealed that plasmid DNA-complexed amino-terminated organically modified silica (ORMOSIL) nanoparticles could transfect neuronal cells after intraventricular injection at amounts comparable to HSV while causing fewer immunological or toxic side effects. It was discovered that nanoparticles were ineffective and only mediated a temporary impact, requiring repeated injections [46].
Vector Targeting By Altering the Capsid
Significant progress has been made in vector targeting with respect to transduction efficiency overall, vector delivery to a broad target region, and occasionally vector delivery to a particular tissue or cell type. Targeting can be made more effective, as demonstrated by the advancements made to AAV vectors in the last ten years. To improve AAV vector targeting, several strategies have been employed, including the use of alternative serotype capsids (the protein shell of a virus), logical mutagenesis of the capsid, insertion of targeting peptides into the capsid, and directed evolution to generate novel capsids [47]. A vast array of vectors for optimal transgene delivery has been made available by the identification of more than 100 AAV capsid variants, each of which may have a distinct cell tropism.
More than a hundred AAV capsid variations have been found; each one may have a distinct cell tropism, offering a wide range of vectors for efficient transgene delivery. For instance, because AAV9 can pass the blood-brain barrier following intravenous injection, it may find value in CNS applications. The high liver tropism of AAV9 (compared to its CNS tropism) could reduce enthusiasm to utilise it as a vector, but this tendency can be decreased by introducing point mutations into the capsid, or by incorporating microRNA target sequences that respond to microRNAs that are highly expressed in the liver but not in the CNS into viral genes or transgenes delivered by viruses to limit their expression in the liver, thereby reducing toxicity to nontarget areas [48].
AAV capsids can be given new properties by peptide insertions. Modified AAV2 capsids that targeted the cerebral vasculature after intravenous injection were developed through the production of unique peptides using a phage display library. Directed evolution can be used with capsid gene shuffles to select for novel features, resulting in completely new AAV capsids.
A unique AAV capsid was developed in one study, and following intravenous injection, it demonstrated enhanced expression at epilepsy-damaged areas and nearly no expression in the liver, heart, or muscle—a good safety and bio distribution profile. Virus capsule engineering has the ability to decrease side effects while increasing vector potency and cell selectivity, much like medicine optimisation [49].
Strategies for Gene Treatment
The development of gene therapy approaches has been demonstrated by the work being conducted on retinal illnesses. The application of gene therapy has been expanded beyond autosomal recessive sensorineural illnesses (Leber congenital amaurosis) to complicated inherited and acquired diseases, such as age-related macular degeneration, through the investigation of at least six transgene transport and expression techniques [50].
Gene augmentation and/or gene knockdown are intended to reduce or eliminate the expression of a hazardous gain-of-function gene product, or to rectify gene expression in the context of a loss-of-function mutation by introducing the wild-type cDNA. Due to the disease’s progressive nature, which frequently results in the therapeutic target cell degenerating and dying, gene augmentation is frequently restricted by a small therapeutic window. Another method of gene therapy is chromosomal correction of the initial genetic damage, although this strategy is difficult due to the low efficacy of existing gene-editing technology. For the treatment of abnormal ocular neovascularization, a vector carrying a transgene encoding a decoy protein is transmitted to the target organ. This strategy involved expressing the sFlt1 transgene, which binds vascular endothelial growth factor, a major pathological cause of ocular neovascularization [51]. The transgenic encodes a tyrosine kinase.
Vectors expressing genes encoding proteins with anti- neovascular or anti-apoptotic characteristics can also be administered. Treatment for choroidal neovascularization, for instance, involves the transfer of the pigment epithelium- derived factor-encoding gene, which has undefined antiangiogenic characteristics.
Nerve regeneration has been improved by upregulating the expression of genes producing growth factors. It has been demonstrated that retinal stem cells can induce a photoreceptor phenotype by expressing the CRX transcription factor. In animal models, this method has also been utilised to generate functioning auditory hair cells in the cochlea. The last possibility for restoring vision is molecular prosthetics, which involves inserting light-sensitive ion channel or ion pump proteins into the retina that are derived from bacteria and algae. Examples of these proteins are the channel Rhodopsin or Halorhodopsin subfamilies [52].
All of the above-discussed methods necessitate some degree of remaining CNS function or enough plasticity to integrate the neural signal from the peripheral organs that have been treated. According to a functional MRI study conducted on patients receiving retinal pigment epilethium- specific 65kDa protein (RPE65) retinal gene augmentation therapy, there is sufficient plasticity in at least some retinal degenerative diseases. These patients showed light-induced cortical responses even after they had been blinded for more than 3.5 decades [53].
Goals of Gene Therapy
PNS and Sensory Organs: Gene therapy for sensorineural disorders has made significant advancements, particularly in the area of severe retinal degenerative diseases, for which there are now no effective treatments. Since the mammalian eye is easily accessible, its immune system responds well to gene transfer, and noninvasive functional and structural investigations are readily available, it has been the target organ in several gene therapy therapeutic trials. Many of these studies have concentrated on rare diseases, such as choroideraemia, Leber congenital amaurosis, and retinitis pigmentosa. These conditions may serve as stepping stones for treating more common blinding conditions like glaucoma, age-related macular degeneration, and diabetic retinopathy, which have few available treatment options [54]. RPE65 gene augmentation has been shown to be safe and effective in treating patients with Leber congenital amaurosis in three separate clinical trials. This approach was made possible by advancements in the identification and cloning of the genes linked to the condition, and it has sparked a number of studies into gene therapy for other hereditary types of blindness. Following the discovery of the first two, more than 25 genes linked to blindness have been identified; these genes can also result in hearing loss and/or vestibular abnormalities (Usher syndrome). The development of animal models of blindness and an enhanced comprehension of disease pathophysiology are results of advancements in the science of genetics [55].
Gene therapy has advanced for additional extraocular sensory impairments, though more slowly than for retinal applications. There is a great need for clinical care since presbycusis is becoming a more common cause of hearing loss and deafness as populations age. Although cochlear problems are more difficult to reach surgically than retinal illnesses, gene therapy has shown promise in treating a number of hereditary deafness-causing conditions. In one of these investigations, a transcription factor assumed to be crucial for the development of stereocilia was used to restore cochlear hair cells following noise-induced degeneration [56].
In a different investigation, mice deficient in vesicular glutamate transporter-3 had a gene encoding the enzyme inserted into their cochlea; gene augmentation treatment helped the mice regain some hearing. In a third crucial proof-of-concept study, a mouse model of Usher syndrome was given antisense oligonucleotides to address a splicing problem in one type of the condition. Lastly, despite the fact that target organs like the tongue and nose are easier to access than the retina and cochlea, relatively few research have focused on smell and taste abnormalities. This is likely because these disorders have complex aetiologies and have unfavourable risk-benefit ratios [57].
Gene Therapy in Vivo: Although it was shown decades ago that therapeutic DNA could be successfully delivered in vitro into mammalian cells, in vivo genetic material delivery has been hindered by a variety of factors. Therapeutic nucleic acids are in circulation, but their half-lives in vivo are brief due to nuclease degradation. The selectivity of the blood- brain barrier (BBB), a large population of diverse glial cells, and the requirement of cell-cycle independent introduction of genetic material given that neurons are post-mitotic and do not divide make in vivo targeting of specific organs and cell types even more challenging. Immunogenicity, toxicity, and mutagenesis are additional risks associated with post- viral integration [58].
The potential uses of in vivo gene therapy have been confirmed by a few significant clinical trials. The first in vivo gene editing procedure to replace the dysfunctional iduronate-2-sulfatase (IDS) enzyme was performed on a patient with mucopolysaccharidosis (MPS) type II, often known as Hunter syndrome, as part of the CHAMPIONS Phase 1 and 2 trials. The accumulation of hazardous carbohydrate metabolites throughout the body causes severe debilitation in MPS II patients in the absence of this enzyme.
The enzyme-replacement therapy (ERT) that is now available works well, however after a day of treatment, IDS levels almost completely disappear. A single intravenous infusion of therapeutic DNA to the liver is being investigated in this research using ZFN editing in conjunction with an AAV vector. ZFNs replace a specific region of the albumin gene through specialised targeting, which only permits activation of DNA instructions by liver cells. This precise and permanent integration of the functioning IDS gene makes possible [59].
With the content of the supplied therapeutic genetic material being the only variation, a second clinical trial is targeting exclusively the albumin gene in liver cells using the same technology as above. Factor IX (FIX), a critical clotting protein, disappears in haemophilia B, an uncommon inherited bleeding condition. Through targeted delivery and integration of a functioning F9 gene, patients can generate an endless supply of the clotting protein in their own systems.
Because of the efficacy and safety of both trials, the FDA has designated both as orphan drugs and fast-track trials, resulting in an exciting new age of gene therapy applications in vivo. In contrast to typical viral vectors that provide a danger of insertional mutagenesis that impedes in vivo applications, the outcome of these studies will highlight the significance of gene insertion selectivity when considering the future of sustainable in vivo therapy [60].
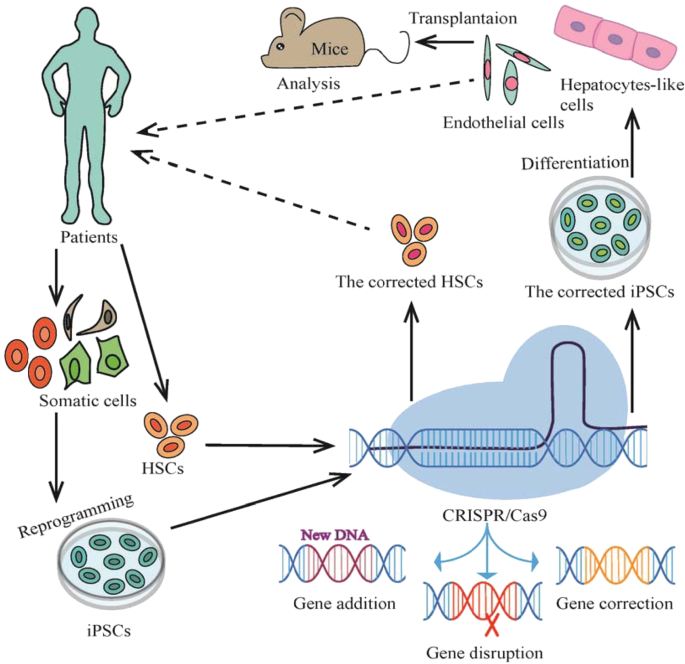
Ex Vivo Gene Transfer Developments: Lentiviral Correction of Bone Marrow Stem Cells: Enzyme replacement therapy (ERT), hematopoietic cell transplantation (HCT), or a combination of the two, as in Hurler’s syndrome, are effective treatment options for lysosomal storage diseases [61]. The primary issue with ERT is that systemically administered enzymes have limited access due to the enzyme’s ability to effectively pass the blood-brain barrier. This can be overcome with allogeneic HCT. The BBB can be crossed by hematopoietic cells, which can be employed to transport enzymes to the central nervous system. Examples of these cells are activated lymphocytes, monocytes, and microglial precursors. After hematopoietic stem cell (HSC) transplantation, cells originating from donors enter the central nervous system (CNS) and undergo differentiation to become parenchymal and perivascular microglia. The cross- correction of the enzyme in nearby neurons and glial cells may be mediated by these cells. It is ineffective for patients with overt neurological or aggressive infantile forms of lysosomal storage disorders (LSDs) and only beneficial for a small proportion of these people. A further drawback of allogeneic HCT is the development of Graft-Vs-Host Disease (GVHD) or Host-Vs-Graft Disease (HVGD), a condition in which the immune system becomes activated and results in significant inflammation. Allogeneic hematopoietic stem cell transplantation (HSCT) issues may be eliminated by using autologous HSCs that have been altered with lentiviral vectors to express the missing protein or enzyme.
Although the spread of the transgene among dividing cells was initially thought to be an advantage of lentiviral vectors, the random insertion of the transgene into the host genome carries the risk of insertional mutagenesis, as demonstrated by the leukaemia that developed in X-linked SCID patients as a side effect of treatment [62]. By minimising or completely removing its integration into the host genome, lentiviral vectors have been made much safer. Lentiviral vector safety is increased and the risk of gene activation is reduced by targeting integration to heterochromatin areas. Other developments in lentiviral vectors include non-integrating lentiviral (NIL) vectors, which carry mutant integrase or mutations in their LTRs that prevent integrase binding, and self-inactivating (SIN) mutations, which eliminate the promoter activity of the LTR. These viral genomes are found episomally in both circular and linear forms. With the use of these innovative technologies, cancer risks are reduced and vector safety is enhanced [63].
Challenges
Numerous crippling diseases may be cured with gene therapy, but its application is now restricted because of significant challenges. Initially, the gene needs to be transferred to the appropriate cells within the appropriate tissue. Adverse iatrogenic effects from improper targeting may occur in healthy tissue, which may amplify germline mutations if, for example, therapeutic genes are unintentionally inserted into a patient’s germline cells. However, advancements in CSF biomarker assessment, brain imaging, and vector design are proposed to increase specificity. The relative absence of CNS biomarkers is an essential barrier to evaluating appropriate cell targeting and pharmacodynamic responses. Second, if the therapy is unable to evade the host’s defences, in vivo administration through the use of a virus vector may cause a strong immunological reaction [64].
Third, the integration of therapeutic genes can interfere with the function of other genes. For example, a gene therapy trial for children with X-linked severe combined immune deficiency (SCID) successfully restored immune function, but in the process, the integration of a gene that is essential for the rate of cell division caused leukaemia in some patients.
Furthermore, there is an unidentified window of opportunity for intervention because many adult-onset neurological illnesses, including AD, may proceed for decades before clinical symptoms show. Moreover, it becomes challenging to find patients who can receive gene therapy sufficiently early in the development of their health condition to benefit from it in any manner. To evaluate the impact of treatment, individuals with quickly progressing monogenic types of ALS, for example, still need to be monitored for a minimum of 12 months [65].
Safety, more than efficacy, is the main barrier to the widespread application of gene therapy. The primary causes of the overexpression of the therapeutic transgene or gene expression in non-target organs are the potential for toxicity and unfavourable iatrogenic effects. For example, methyl- CpG-binding protein 2 (MECP2) gene mutations cause Rett syndrome in females, whereas gene duplications induce intellectual impairment in males, and overexpression in mice models results in aberrant behaviours and seizures. Consequently, the therapeutic transgene expression has limitations to a very specific therapeutic index when treating conditions like Rett syndrome. The degree of additional toxicity is contingent upon the function of endogenous genes and the timing of the intervention in relation to the advancement of the disease. Safety concerns are exacerbated by the incapacity to accurately control in vivo expression [66]. The main concerns are the possibility of mutagenesis after viral integration into the host genome and harmful immunological reactions, particularly when using viral vectors.
A certain amount of expression control is made possible by the demonstrated abilities of RNA interference (RNAi) and micro-RNA (miRNA) to prevent the overexpression of therapeutic genes delivered by viruses. However, there are few clinical trials evaluating the in vivo application of miRNA, and they still fall short of addressing other potential safety concerns related to viral vectors [67].
Lipids, nanoparticles, and polymers are examples of recent developments in the development of non-viral vectors that help reduce adverse responses and have better safety profiles than viral vectors. The issues of target cell specificity, integration, downstream effects, permeability/ bioavailability, variable disease progression between individuals, and timing of administration relative to disease onset still remain fundamental concerns when considering gene therapy as a treatment option, despite numerous successful clinical trials [68, 69, 70, 71, 72, 73, 74, 75].
Conclusion
Gene therapy was first proposed as a science fiction remedy for some diseases, yet over the last several decades, it has become a fact of life. The field of gene-based therapy for neurological conditions such as ALS, spinal muscular atrophy, X-linked adrenoleukodystrophy, AD, ASD, and even cancerous diseases has undergone a revolution because of recent advancements. Despite encouraging findings from research, there are still a number of challenges that need to be overcome. Current challenges that need to be tackled involve the immunogenicity and toxicity of vectors, oncogenic insertions into the genome, and the difficulties of translating from mice to human models. However, given its enormous therapeutic promise, gene therapy for neurological diseases offers interesting new treatments. Overcoming these challenges will be crucial to bringing gene therapy for neurological disorders from bench to the bedside. Clinical experiments conducted in the previous ten years have demonstrated beneficial results of gene transfer, however the benefit to the patients has been minimal. In recent years, there has been a tremendous advancement in vector technology for both in vivo and ex vivo gene transfer. Novel methodologies have been developed to improve the efficiency and reach of gene transfer. The preclinical animal experiments are demonstrating the efficacy of these techniques for in vivo and ex vivo gene transfer, and they are in an advantageous circumstance to move quickly into clinical trials. Even small clinical achievements will give rise to hope and propel the field ahead. If the current preclinical accomplishments are translated into the clinical setting, the upcoming years should be highly captivating.
Key Points
The majority of PNS and CNS diseases cannot be adequately treated with standard pharmaceutical and surgical interventions.
- Gene therapy is a viable strategy to prevent the progression of neurological diseases and may even be able to slow down the disease process.
- The effectiveness of gene therapy depends on the development of gene delivery vehicles, primarily viral vectors, to deliver disease-modifying products to the necessary locations.
- Treatment strategies for some debilitating pain and vision disorders, as well as diseases affecting vision and hearing, are advanced in development.
- Gene therapy for degenerative diseases necessitates a deeper comprehension of the underlying pathophysiology and, in some cases, global brain delivery of the transgene.
- Patients should have access to these medicines within ten years due to the increasing development of gene therapy applications for diseases of the nervous system.
References
-
Baxter LR, Schwartz JM, Phelps ME, Mazziotta JC, Guze BH, et al. (1989) Reduction of prefrontal cortex glucose metabolism common to three types of depression. Archives of general psychiatry **46(3): 243-** 250.
-
Sayed N, Allawadhi P, Khurana A, Singh V, Navik U, et al. (2022) Gene therapy: Comprehensive overview and therapeutic applications. Life sciences **294**: 120375.
-
Mueller C, Flotte TR (2008) Clinical gene therapy using recombinant adeno-associated virus vectors. Gene therapy 15(11): 858-863.
-
Romano G, Pacilio C, Giordano A (1999) Gene transfer technology in therapy: current applications and future goals. Stem cells 17(4): 191-202.
-
Lin CY, Hsieh HY, Chen CM, Wu SR, Tsai CH, et al. (2016) Non-invasive, neuron-specific gene therapy by focused ultrasound-induced blood-brain barrier opening in Parkinson’s disease mouse model. Journal of Controlled Release 235: 72-81.
-
Kim SU, De Vellis J (2009) Stem cell‐based cell therapy in neurological diseases: a review. Journal of neuroscience research 87(10): 2183-2200.
-
Bassett DS, Gazzaniga MS (2011) Understanding complexity in the human brain. Trends in cognitive sciences 15(5): 200-209.
-
Cornford EM, Hyman S (1999) Blood–brain barrier permeability to small and large molecules. Advanced drug delivery reviews 36(2-3): 145-163.
-
Pan W, Banks WA, Kastin AJ (1998) Permeability of the blood–brain barrier to neurotrophins. Brain research 788(1-2): 87-94.
-
Abbott NJ (2000) Inflammatory mediators and modulation of blood–brain barrier permeability. Cellular and molecular neurobiology 20(2): 131-147.
-
Gaillard PJ, de Boer AG (2000) Relationship between permeability status of the blood–brain barrier and in vitro permeability coefficient of a drug. European journal of pharmaceutical sciences 12(2): 95-102.
-
Miyake MM, Bleier BS (2015) The blood-brain barrier and nasal drug delivery to the central nervous system. American journal of rhinology & allergy 29(2): 124-127.
-
Bellettato CM, Scarpa M (2018) Possible strategies to cross the blood–brain barrier. Italian journal of paediatrics 44(2): 127-133.
-
Kimura S, Harashima H (2020) Current status and challenges associated with CNS-targeted gene delivery across the BBB. Pharmaceutics 12(12): 1216.
-
Timbie KF, Mead BP, Price RJ (2015) Drug and gene delivery across the blood–brain barrier with focused ultrasound. Journal of Controlled Release 219: 61-75.
-
Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, et al. (2013) Progress in gene therapy for neurological disorders. Nature Reviews Neurology 9(5): 277-291.
-
Iqubal A, Iqubal MK, Khan A, Ali J, Baboota S, et al. (2020) Gene therapy, a novel therapeutic tool for neurological disorders: current progress, challenges and future prospective. Current Gene Therapy 20(3): 184-194.
-
Tuszynski, MH (2002) Growth-factor gene therapy for neurodegenerative disorders. The Lancet Neurology 1(1): 51-57.
-
Piguet F, de Saint Denis T, Audouard E, Beccaria K, André A, et al. (2021) The challenge of gene therapy for neurological diseases: strategies and tools to achieve efficient delivery to the central nervous system. Human Gene Therapy 32(7-8): 349-374.
-
Wood MJ, Trülzsch B, Abdelgany A, Beeson D (2003) Therapeutic gene silencing in the nervous system. Human molecular genetics 12(2): R279-R284.
-
Nóbrega C, Mendonça L, Matos CA (2020) Gene Therapy Strategies: Gene Silencing. A Handbook of Gene and Cell Therapy, pp: 127-146.
-
Wykes R, Lignani G (2018) Gene therapy and editing: Novel potential treatments for neuronal channelopathies. Neuropharmacology 132: 108-117.
-
McMahon M, Cleveland D (2017) Gene therapy: Gene- editing therapy for neurological disease. Nature Reviews Neurology 13: 7-9.
-
Madigan N, Staff N, Windebank A, Benarroch E (2017) Genome editing technologies and their potential to treat neurologic disease. Neurology 89(16): 1739 - 1748.
-
Day JJ (2019) Genetic and epigenetic editing in nervous system. Dialogues in Clinical Neuroscience 21(4): 359- 368.
-
Sinnamon J, Kim S, Fisk J, Song Z, Nakai H, et al. (2020) In Vivo Repair of a Protein Underlying a Neurological Disorder by Programmable RNA Editing. Cell reports 32(3): 107878.
-
Kay M, Glorioso J, Naldini L (2001) Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nature Medicine 7(1): 33-40.
-
Waehler R, Russell S, Curiel D (2007) Engineering targeted viral vectors for gene therapy. Nature Reviews Genetics 8: 573-587.
-
Kotterman M, Chalberg T, Schaffer D (2015) Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annual review of biomedical engineering 17: 63-89.
-
Shirley J, Jong Y, Terhorst C, Herzog R (2020) Immune Responses to Viral Gene Therapy Vectors. Molecular Therapy 28(3): 709-722.
-
Lundstrom K (2003) Latest development in viral vectors for gene therapy. Trends in biotechnology 21(3): 117- 122.
-
Robbins P, Tahara H, Ghivizzani S (1998) Viral vectors for gene therapy. Trends in biotechnology 16(1): 35-40.
-
Wang D, Tai P, Gao G (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery 18: 358-378.
-
Kochanek S, Clemens P, Mitani K, Chen H, Chan S, et al. (1996) A new adenoviral vector: Replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta- galactosidase.. Proceedings of the National Academy of Sciences of the United States of America 93(12): 5731- 5736.
-
Zufferey R, Nagy D, Mandel R, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnology 15: 871- 875.
-
Kafri T, Blömer U, Peterson D, Gage F, Verma I (1997) Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nature Genetics 17(3): 314-317.
-
Follenzi A, Ailles L, Bakovic S, Geuna M, Naldini L (2000) Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nature Genetics 25(2): 217-222.
-
Amado R, Chen I (1999) Lentiviral Vectors--the Promise of Gene Therapy Within Reach?. Science 285(5428): 674-676.
-
Wang G, Levine B, Binder G, Berry C, Malani N, et al. (2009) Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Molecular therapy: the journal of the American Society of Gene Therapy 17(5): 844-850.
-
Yin H, Kanasty R, Eltoukhy A, Vegas A, Dorkin J, et al. (2014) Nature Reviews Genetics 15: 541-555.
-
Naldini L (2009) A Comeback for Gene Therapy. Science 326(5954): 805-806.
-
Lv H, Zhang S, Wang B, Cui S, Yan J (2006) Toxicity of cationic lipids and cationic polymers in gene delivery. Journal of controlled release: official journal of the Controlled Release Society 114(1): 100-109.
-
Wasungu L, Hoekstra D (2006) Cationic lipids, lipoplexes and intracellular delivery of genes. Journal of controlled release: official journal of the Controlled Release Society 116(2): 255-264.
-
Ar**ís** A, Villaverde A (2004) Modular protein engineering for non-viral gene therapy. Trends in biotechnology 22(7): 371-377.
-
Liu Y, Yin L (2020) **α-Amino acid N-**carboxyanhydride (NCA)-derived synthetic polypeptides for nucleic acids delivery. Advanced drug delivery reviews 171: 139-163.
-
Cullis P, Hope M (2017) Lipid Nanoparticle Systems for Enabling Gene Therapies. Molecular therapy: the journal of the American Society of Gene Therapy 25(7): 1467- 1475.
-
Fuente M, Seijo B, Alonso M (2008) Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Investigative ophthalmology & visual science 49(5): 2016-2024.
-
Liu G, Swierczewska M, Lee S, Chen X (2010) Functional Nanoparticles for Molecular Imaging Guided Gene Delivery. Nano today 5 (6): 524-539.
-
Roth J, Cristiano R (1997) Gene therapy for cancer: what have we done and where are we going?. Journal of the National Cancer Institute 89(1): 21-39.
-
Nettelbeck D, Jérôme V, Müller R (2000) Gene therapy: designer promoters for tumour targeting. Trends in genetics 16(4): 174-181.
-
Arruda V, Favaro P, Finn J (2009) Strategies to modulate immune responses: a new frontier for gene therapy.. Molecular therapy: the journal of the American Society of Gene Therapy 17(9): 1492-503.
-
Avalosse B, Dupont F, Burny A (1995) Gene therapy for cancer. Current opinion in oncology 7(1): 94-100.
-
Cesur-Ergün B, Demir-Dora D (2023) Gene therapy in cancer. The Journal of Gene Medicine 25(11): e3550.
-
Menon A, Eb M, Kuppen P, Velde C (2002) Gene Therapy Strategies for Colorectal Cancer. Colorectal Cancer, pp: 811-835.
-
Abaan OD, Criss WE (2002) Gene Therapy in Human Breast Cancer. Turkish Journal of Medical Sciences 32: 283-291.
-
Yin H, Kanasty R, Eltoukhy A, Vegas A, Dorkin J, et al. (2014) Non-viral vectors for gene-based therapy. Nature Reviews Genetics 15: 541-555.
-
Culver KW (1994) Clinical applications of gene therapy for cancer. Clinical chemistry 40(4): 510-512.
-
Lafont A, Guérot C, Lemarchand P (1996) Prospects for gene therapy in cardiovascular disease. European heart journal 17(9): 1312-1317.
-
Cantore A, Fraldi A, Meneghini V, Gritti A (2022) In vivo Gene Therapy to the Liver and Nervous System: Promises and Challenges. Frontiers in Medicine 8: 774618.
-
Gregory-Evans K, Bashar A, Tan M (2012) Ex-vivo gene therapy and vision. Current gene therapy 12(2): 103- 115.
-
Vogel JC (1999) A direct in vivo approach for skin gene therapy. Proceedings of the Association of American Physicians 111 (3): 190-197.
-
Raymon H, Thode S, Gage F (1997) Application ofex VivoGene Therapy in the Treatment of Parkinson’s Disease. Experimental Neurology 144(1): 82-91.
-
Scherer L, Rossi J (2011) Ex vivo gene therapy for HIV-1 treatment. Human molecular genetics, 20(R1): R100-R107.
-
Grossman M, Raper S, Kozarsky K, Stein E, Engelhardt J, et al. (1994) Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nature Genetics 6(4): 335-341.
-
Ishida A, Yasuzumi F (2000) Approach to ex vivo gene therapy in the treatment of Parkinson’s disease. Brain & development 22(1): S143-S147.
-
Gowing G, Svendsen S, Svendsen C (2017) Ex vivo gene therapy for the treatment of neurological disorders. Progress in brain research 230: 99-132.
-
Verma I, Somia N (1997) Gene therapy - promises, problems and prospects. Nature 389: 239-242.
-
Pena SA, Iyengar R, Eshraghi RS, Bencie N, Mittal J, et al. (2020) Gene therapy for neurological disorders: challenges and recent advancements. J Drug Target 28(2): 111-128.
-
Morris G, Schorge S (2022) Gene Therapy for Neurological Disease: State of the Art and Opportunities for Next- generation Approaches. Neuroscience 490: 309-314.
-
Brenner D, Ludolph AC, Weishaupt JH (2020) Gene specific therapies – the next therapeutic milestone in neurology. Neurol. Res. Pract 8: 2-25.
-
Paul A, Collins MG, Lee HY (2022) Gene Therapy: The Next-Generation Therapeutics and Their Delivery Approaches for Neurological Disorders. Front. Genome Ed 4: 899209.
-
Martins IJ (2016) Anti-Aging Genes Improve Appetite Regulation and Reverse Cell Senescence and Apoptosis in Global Populations. Advances in Aging Research 5(1): 9-26.
-
Martins IJ (2017) Single Gene Inactivation with Implications to Diabetes and Multiple Organ Dysfunction Syndrome. J Clin Epigenet 3(3): 24.
-
Martins IJ (2019) Appetite Control and Core Body Temperature are linked to Autoimmune Disease and the Global Chronic Disease Epidemic. Enliven: Immunol Immunotechnol 3(e1): 001-009.
-
Martins IJ (2017) Nutrition Therapy Regulates Caffeine Metabolism with Relevance to NAFLD and Induction of Type 3 Diabetes. J Diabetes Metab Disord 4: 019.
- Grandma, Don’t Forget How Much I Love You: A Teaching and Learning Resource Embedded in the Rich Jamaican Culture
- Current Pharmacotherapeutic Approaches for Management of Alzheimer’s Disease
- Evidence-Based Guideline: Individualized Music for Persons with Dementia (7th Edition) — Non-Pharmacological Intervention for Agitation
- Alzheimer’s Disease: Invisible Friend of Old People
- Psychological Needs of the Population in Saint Petersburg in Time of the Covid-19 Pandemic
- Amyloid B-Protein Aggregation at Physiologically Relevant Concentrations. A Critical Role of Membranes