Bioequivalence and Pharmacokinetics of Carvedilol (6.25 and 12.5 Mg Tablets) in Healthy Thai Volunteers
The Government Pharmaceutical Organization (GPO) had developed the generic products of carvedilol 12.5 and 6.25 mg tablets as low-cost alternatives for patients and physicians to enhance patient adherence and accessibility to long-term use medications. Two bioequivalence studies were conducted to evaluate the bioequivalence between the test and reference products of carvedilol 12.5 mg and 6.25 tablets. The design for both studies was comparative randomized, open-label, singledose, two-way crossover. Carvedilol and 4′-hydroxyphenyl carvedilol (active metabolite) concentrations in plasma were simultaneously determined by a validated liquid chromatography tandem mass spectrometry method. The pharmacokinetic analysis was carried out for carvedilol and 4′-hydroxyphenyl carvedilol. The pharmacokinetic parameters describing the rate and extent of absorption of carvedilol (AUC0-tlast, AUC0-∞ and Cmax) were used to conclude the bioequivalence between the test and reference products whereas the pharmacokinetic parameters of 4′-hydroxyphenyl carvedilol were presented as supportive information. The statistical analysis was calculated using an analysis of variance and did not show any significant difference between the two formulations. The 90% confidence intervals of the geometric least squares mean ratios (test/ reference) of ln-transformed AUC0-tlast, AUC0-∞ and Cmax were 98.54-106.94%, 98.54-106.56% and 90.70-104.84%, respectively for carvedilol 12.5 mg tablets, and 95.56-105.99%, 94.80-104.98% and 92.00-107.33%, respectively for carvedilol 6.25 mg tablets. The results were well within 80.00-125.00% corresponding to the bioequivalence criteria. Therefore, the generic carvedilol products (Carvolol) at both strengths are bioequivalent with the innovator products (Dilatrend) in terms of rate and extent of absorption. Similar findings were observed for 4′-hydroxyphenyl carvedilol, thus therapeutic equivalence between the test and reference formulations could also be anticipated. The products were well tolerated by the study subjects and no serious adverse events were reported in both studies.
Introduction
Hypertension is a risk factor leading to several cardiovascular diseases, for example, congestive heart failure, angina pectoris and cardiac arrhythmias. In Thailand, uncontrolled hypertension is associated with several intrinsic and extrinsic factors including the number of antihypertensive medications. The data indicated that hypertensive patients trended to receive multiple medications to control blood pressure as well as to prevent serious consequences [1]. Since the patients need to use the medications continuously, the development of generic products could be one of the solutions to enhance patient adherence and accessibility to long-term use medications.
β-blocking agents such as metoprolol, atenolol or carvedilol are a class of drug exerting the activity via blockade the binding of neurotransmitters, e.g., epinephrine and norepinephrine to β-adrenergic receptor [2]. Carvedilol is a third generation, nonselective β-blocker used to treat hypertension, chronic heart failure, angina pectoris and cardiac arrhythmias [3, 4]. Carvedilol is rapidly absorbed with peak plasma concentration attained at approximately 1 hour following oral administration. Carvedilol is primarily metabolized to 4′-hydroxyphenyl carvedilol and 5′-hydroxyphenyl carvedilol by cytochrome P450 enzymes expressed in liver accounted for about 75% of the administered dose [5]. The pre-systemic metabolism of carvedilol is stereoselective which S (-)-carvedilol is extensively metabolized resulting in three-fold lower plasma concentration of S (-)-carvedilol than R (+)-carvedilol [6, 7]. The 4′-hydroxyphenyl metabolite is more potent than S (-)-carvedilol for β-adrenoreceptor blocking activity [5], while R (+)-carvedilol contributes to α1-adrenoreceptor blocking activity responsible for vasodilatory property [6, 7, 8]. In addition, carvedilol also exhibits antioxidative and antiproliferative properties that would be beneficial for patients with cardiovascular comorbidity [9].
The Government Pharmaceutical Organization (GPO) had developed the generic products of carvedilol 12.5 and 6.25 mg tablets as low-cost alternatives for patients and physicians. Because of the difference in the formulations of generic carvedilol tablets between both strengths, two bioequivalence studies were conducted to compare the pharmacokinetic parameters describing the rate and extent of absorption between the test (Carvolol 12.5 mg and 6.25 mg tablets, GPO) and reference (Dilatrend 12.5 mg and 6.25 mg tablets, Roche S.p.A. Milan. Segrate) formulations at each strength individually. The bioequivalence studies would provide supportive information on the interchangeability between generic and innovator formulations, as well as to evaluate the safety of the formulations in healthy Thai volunteers for generic drug registration in Thailand.
Materials and Method
Study Products
The test products of Carvedilol 6.25 mg and 12.5 mg tablets (Carvolol) were manufactured by the Government Pharmaceutical Organization, Thailand bearing lot No. S590075 and S600017, respectively. The reference products, Dilatrend 6.25 mg and 12.5 mg tablets were manufactured by Roche S.p.A. Milan. Segrate, Italy bearing lot No. M2099B05 and M2042B09, respectively.
Study Subjects
The number of subjects was estimated based on the intra-subject coefficient of variation (CV) of previous bioequivalence study of carvedilol [10]. The intra-subject CV was included in the estimation along with the estimated values of geometric least squares mean ratio (test/reference) of 0.95, significant level at 0.05 and bioequivalence range of 80.00-125.00%. Following the estimation, 51 subjects was sufficient for establishing bioequivalence with the power greater than 0.8. Total of 62 subjects was recruited in both studies to compensate 20% dropouts and withdrawals.
In each study, 62 healthy Thai male and female subjects with the age between 18-55 years and the body mass index (BMI) between 18 and 25 kg/m2 were enrolled. All subjects were divided into two groups according to a randomization schedule generated using SAS version 9.3 (SAS Institute Inc., USA). The subjects were screened through medical history, demographic data, physical examination (vital signs, 12-Lead ECG and chest X-ray) and laboratory tests (haematology, biochemistry, virology, urinalysis and breath test for alcohol consumption) prior to dosing in period I. Vital signs (blood pressure, pulse rate, body temperature and respiratory rate) were checked periodically in each period. Laboratory tests and 12-Lead ECG were also performed at the end of study. Subjects were monitored throughout the study period for any adverse events regardless of its association with the study medication. The severity of the adverse event was determined as mild, moderate, or severe.
Study Design
Two separate comparative randomized, open-label, single-dose, two-way crossover bioequivalence studies of carvedilol 12.5 mg and 6.25 mg tablets were conducted at International Bio Service Co., Ltd., Golden Jubilee Medical
Center, Mahidol University, Thailand. In first period, either a tablet of test or reference product was orally administered to each subject after an overnight fasting. Subjects were received an alternate treatment after a 7-day washout period. In each period, no water except the 240 mL given with drug administration was allowed within 1 hour before and after dosing. Meals were served at 4 and 10 hours after dosing for lunch and dinner, respectively. Taking any concomitant medications, vitamins, or dietary supplements were restricted for 14 days before the study and entire duration of the study. The study protocols were approved by Institute for the Development of Human Research Protections (IHRP), Department of Medical Sciences, Ministry of Public Health, Thailand in accordance with the Declaration of Helsinki and its amendments [11] and the International Conference on Harmonization Guideline for Good Clinical Practice [12].
Blood Sampling
Around 5 mL of Blood samples (~7 mL for a pre-dose sample) were collected through an indwelling intravenous cannula placed in a forearm vein of the subjects. Blood samples were transferred into K2EDTA-containing tubes and sample tubes were placed in a wet ice water bath until centrifugation. Blood samples were drawn at pre-dose (0 hour) and 0.083, 0.167, 0.25, 0.333, 0.5, 0.667, 0.833, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 16, 24, 36 and 48 hours post-dose. The blood samples were centrifuged at 3000 ± 100 rcf, below 10°C for 5 minutes to separate plasma. All separated plasma samples were transferred to pre-labelled polypropylene tubes and stored at -55°C or colder until sample analysis.
Study Sample Analysis
The plasma concentrations of carvedilol and 4′-hydroxyphenyl carvedilol were simultaneously analysed using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) bioanalytical method over concentration ranges of 99.842 to 99932.440 pg/mL and 50.461 to 10101.350 pg/mL, respectively. For sample preparation, 500 μL of plasma was processed using liquid- liquid extraction technique by adding 0.02 M sodium hydroxide solution and diethyl ether: hexane (8:2) mixture into each sample. Carvedilol-d5 and 4′-hydroxyphenyl Carvedilol-d5 were used as internal standards for carvedilol and 4′-hydroxyphenyl carvedilol, respectively. The samples were centrifuged and flash-frozen to separate the organic layer. The contents were evaporated and reconstituted with acetonitrile: 0.01% formic acid solution (v/v) (1:1). Subsequently, 8 μL of a reconstituted sample was injected into a chromatographic system consisting of an ACE 5 C18 100×4.6 mm column (Advanced Chromatography Technologies Ltd, Aberdeen, Scotland), a Nexera UPLC system (Shimadzu Corporation, Kyoto, Japan) and a TSQ Quantum Ultra mass spectrometer (Thermo Fisher scientific, Massachusetts, USA). A gradient elution by changing composition of 0.01 % Formic acid solution (v/v) and acetonitrile at a flow rate of 0.8 mL/min was used. Mass spectrometry analysis was conducted in positive ionization mode using a multiple monitoring reaction at mass-to-charge ratio (m/z) transitions of 407.146→100.180 for carvedilol and 423.145→100.180 for 4′-hydroxyphenyl carvedilol. The spray voltage was set at 4000 Volts and the capillary temperature was set at 350°C. The vaporizer temperature was set at 450°C with sheath gas and aux gas pressures at 50 psi and 25 psi, respectively. All data acquisitions and processing were accomplished using LCquan version 2.9.0.34 (Thermo Fisher scientific, Massachusetts, USA). The samples were analyzed as per in- house standard operating procedures of GPO complying with Guideline on bioanalytical method validation of European Medicines Agency (EMA) [13] and the U.S. FDA Guidance for Industry on Bioanalytical Method Validation [14].
Pharmacokinetic and Statistical Analysis
The pharmacokinetic parameters were calculated by non- compartmental analysis using Phoenix WinNonlin Software Version 6.3 (Pharsight Corporation, USA). The maximum concentration (Cmax) and time at maximum concentration (tmax) were obtained directly from the pharmacokinetic profile. The area under the plasma concentration-time curve from time zero to the last observed time point (AUC0-tlast) was calculated by linear trapezoidal method. The area under the plasma concentration-time curve from time zero to infinity (AUC0-∞) was calculated from an equation AUC0-tlast + Ct/λz, where Ct was the last observed concentration and λz was the terminal elimination rate constant calculated from the slope of the linear regression of the ln-transformed of the plasma concentration-time curve. The ln-transformed AUC0-tlast, AUC0-∞ and Cmax of carvedilol were primary pharmacokinetic parameters used to determine the bioequivalence between the test and reference products. In contrast, tmax, half-life (t1/2) and percent AUC extrapolation from tlast to infinity of carvedilol were reported as secondary pharmacokinetic parameters. All pharmacokinetic parameters of 4′-hydroxyphenyl carvedilol were presented as supportive information.
The statistical analysis was carried out using SAS® Version 9.3 (SAS Institute Inc., USA). Analysis of variance (ANOVA) was performed for ln-transformed AUC0-tlast, AUC0-∞ and Cmax of carvedilol and 4′-hydroxyphenyl carvedilol running through a general linear model (GLM) of SAS. ANOVA model included sequence, formulation and period as fixed effects and subject nested within sequence, subject (sequence) as a random effect. Sequence effect was tested using subject (sequence) as an error term. Wilcoxon signed rank test was performed to assess the difference in tmax of carvedilol and
4′-hydroxyphenyl carvedilol between test and reference products. If the 90% confidence interval (CI) for the ratios of geometric least squares mean fell within 80.00-125.00% for ln-transformed AUC0-tlast, AUC0-∞ and Cmax of carvedilol, bioequivalence between the test and reference was to be concluded.
Results
Demographic Data
Sixty-two healthy Thai subjects were enrolled in the bioequivalence study of carvedilol 12.5 mg tablets, from which two subjects were withdrawn due to orthostatic hypotension and one subject dropped out due to personal reason. Similarly, the bioequivalence study of carvedilol 6.25 mg tablets was also conducted in 62 healthy Thai subjects, and one subject dropped out due to personal reason. The demographic data of healthy Thai subjects from both studies are summarized in (Table 1).
| Carvedilol 12.5 mg tablets (n=62) | Carvedilol 6.25 mg tablets (n=62) | |
|---|---|---|
| Age (years) | 33.63 (10.47) | 31.65 (7.45) |
| Weight (kg) | 58.97 (8.45) | 60.60 (8.98) |
| Height (m) | 1.63 (0.08) | 1.65 (0.09) |
| BMI (kg/m²) | 22.05 (2.02) | 22.16 (1.95) |
Table 1: Baseline demographic characteristics of enrolled subjects. Data are presented as Mean (SD).
Study Sample Analysis
Total of 3,019 samples from the bioequivalence study of carvedilol 12.5 mg tablets were well received and analysed at the analytical facility of GPO. There were 1.5% and 8.3% of reanalysed samples for carvedilol and 4′-hydroxyphenyl carvedilol, respectively. On the other hand, total 3,075 samples were collected and analysed for the bioequivalence study of carvedilol 6.25 mg tablets. There were less than 3% of repeat analysis for both carvedilol and 4′-hydroxyphenyl carvedilol. In both studies, the quality control samples for both carvedilol and 4′-hydroxyphenyl carvedilol analysed along with the study samples also demonstrated good precision and accuracy during the analysis. The precision of the quality control samples was less than 10% CV for carvedilol and 4′-hydroxyphenyl carvedilol. The accuracy was within ±10% of the nominal concentrations.
Pharmacokinetic and Statistical Analysis
According to the data, carvedilol was rapidly absorbed with the mean Cmax around 60 ng/mL and 30 ng/mL after oral administration of carvedilol 12.5 mg and 6.25 mg tablets, respectively which was attained within 2 hours. The mean AUC0-∞ increased from approximately 100 ng.h/mL to 200 ng.h/mL with the increased dose. In contrast, the mean Cmax of 4′-hydroxyphenyl carvedilol around 10 ng/mL and 4 ng/mL was observed after oral administration of carvedilol 12.5 mg and 6.25 mg tablets, respectively. The median tmax values of carvedilol and 4′-hydroxyphenyl carvedilol were similar. These findings were similar between the test and reference products at both strengths (Table 2).
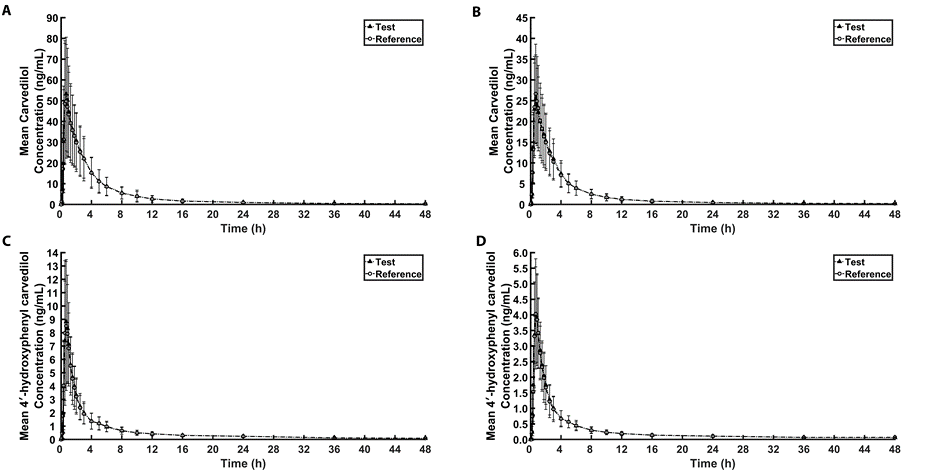
The mean plasma concentration-time profiles of two formulations of carvedilol 12.5 mg tablets obtained from 59 healthy Thai subjects who completed the study are shown in Fig. 1(A) for carvedilol and Fig. 1(C) for 4′-hydroxyphenyl carvedilol. The pharmacokinetic parameters are presented in (Table 2).
| Parameter (Unit) | Carvedilol 12.5 mg tablets (n=59) | Carvedilol 6.25 mg tablets (n=61) | ||
|---|---|---|---|---|
| Test | Reference | Test | Reference | |
| Carvedilol | ||||
| AUC (ng.h/mL) 0-tlast | 201.4 (89.4) | 196.8 (90.0) | 94.4 (37.6) | 94.9 (39.6) |
| AUC (ng.h/mL) 0-∞ | 205.9 (91.6) | 201.7 (92.1) | 97.4 (38.703) | 98.7 (40.9) |
| C (ng/mL) max | 58.4 (28.5) | 58.9 (25.1) | 28.0 (10.3) | 29.1 (12.1) |
| t (h)* max | 0.667 (0.333,2.000) | 0.667 (0.333,1.750) | 0.667 (0.333,2.000) | 0.667 (0.333,1.750) |
| λ (1/h) z | 0.081 (0.034) | 0.076 (0.032) | 0.082 (0.043) | 0.081 (0.045) |
| t (h) 1/2 | 10.3 (4.67) | 10.6 (4.18) | 10.5 (5.05) | 11.2 (6.53) |
| AUC Extrapolation (%) | 2.23 (2.02) | 2.40 (2.03) | 3.09 (1.71) | 3.90 (2.89) |
| 4′-hydroxyphenyl carvedilol | ||||
| AUC (ng.h/mL) 0-tlast | 26.8 (8.29) | 26.6 (8.76) | 12.0 (3.38) | 12.0 (3.83) |
| AUC (ng.h/mL) 0-∞ | 28.4 (8.51) | 28.8 (9.48) | 14.0 (6.05) | 13.4 (4.15) |
| C (ng/mL) max | 9.58 (4.68) | 9.92 (4.72) | 4.37 (1.42) | 4.34 (1.71) |
| t (h)* max | 0.667 (0.500,1.750) | 0.667 (0.500,2.000) | 0.667 (0.500,2.000) | 0.667 (0.500,1.750) |
| λ (1 / h) z | 0.060 (0.020) | 0.054 (0.019) | 0.069 (0.034) | 0.071 (0.035) |
| t (h) 1/2 | 12.7 (3.71) | 14.5 (6.71) | 15.1 (26.0) | 12.1 (5.98) |
| AUC Extrapolation (%) | 5.87 (2.81) | 7.55 (5.88) | 11.13 (10.25) | 10.73 (5.99) |
Table 2: Pharmacokinetic parameters of carvedilol and 4′-hydroxyphenyl carvedilol for the test (Carvolol) and reference (Dilatren
The 90% CIs for the ratios of geometric least squares mean of ln-transformed AUC0-tlast, AUC0-∞ and Cmax of carvedilol were 98.54-106.94%, 98.54-106.56% and 90.70-104.84%, respectively which were within the acceptance range of 80.00-125.00% (Table 3).
| Parameters | Carvedilol 12.5 mg tablets (n=59) | Carvedilol 6.25 mg tablets (n=61) | ||
|---|---|---|---|---|
| Ratio of geometric least squares mean (Test/Reference) | 90% CI | Ratio of geometric least squares mean (Test/Reference) | 90% CI | |
| Carvedilol | ||||
| ln AUC 0-tlast | 102.70% | 98.54-106.94% | 100.60% | 95.56-105.99% |
| ln AUC 0-∞ | 102.50% | 98.54-106.56% | 99.80% | 94.80-104.98% |
| ln C max | 97.50% | 90.70-104.84% | 99.40% | 92.00-107.33% |
| 4′-hydroxyphenyl carvedilol | ||||
| ln AUC 0-tlast | 101.60% | 97.67-105.60% | 101.10% | 95.97-106.58% |
| ln AUC 0-∞ | 99.50% | 95.65-103.55% | 102.90% | 96.07-110.16% |
| ln C max | 96.40% | 89.72-103.58% | 102.90% | 96.32-109.86% |
Table 3: Statistical comparison of ln-transformed primary pharmacokinetic parameters of carvedilol and 4′-hydroxyphenyl carvedilo
For the bioequivalence study of carvedilol 6.25 mg tablets, the mean plasma concentration-time profiles of carvedilol and 4′-hydroxyphenyl carvedilol obtained from 61 healthy Thai subjects who completed the study are shown in Fig. 1(B) and Fig. 1(D), respectively. The pharmacokinetic parameters are presented in Table 2. The 90% CIs for the ratios of geometric least squares mean of ln-transformed AUC0-tlast, AUC0-∞ and Cmax of carvedilol were 95.56-105.99%, 94.80-104.98% and 92.00-107.33%, respectively, which were well within the 80.00-125.00% range (Table 3).
Furthermore, the pharmacokinetics of 4′-hydroxyphenyl carvedilol was characterized and presented in Table 2. Although it was provided as supportive information, the statistical comparison performed for both bioequivalence studies suggested insignificant difference in primary pharmacokinetic parameters of 4′-hydroxyphenyl carvedilol between the test and reference formulations. In both studies, ANOVA model showed no statistically significant effects of sequence, formulation and period on any primary pharmacokinetic parameters of carvedilol and 4′-hydroxyphenyl carvedilol (p>0.05). Wilcoxon signed rank test suggested that there was no significant difference in tmax of carvedilol and 4′-hydroxyphenyl carvedilol between the test and reference products for both studies (p>0.05).
Tolerability
Forty-five adverse events were reported in 29 subjects in the bioequivalence study of carvedilol 12.5 mg tablets. However, the incidence of adverse events was lower for the bioequivalence study of carvedilol 6.25 mg tablets, in which total of 10 adverse events were reported in 9 subjects. The commonly reported adverse events in both studies were headache and dizziness whereas vascular disorders including asymptomatic hypotension and orthostatic hypotension were more frequently found in the bioequivalence study of carvedilol 12.5 mg tablets. No serious adverse events were reported after the study drug administration. All adverse events were summarized in (Table 4).
| Adverse event | Incidence | |||
|---|---|---|---|---|
| Carvedilol 12.5 mg tablets | Carvedilol 6.25 mg tablets | |||
| Test | Reference | Test | Reference | |
| Bradycardia | 2 | 3 | 1 | 0 |
| Palpitation | 0 | 1 | 0 | 0 |
| ALT increased | 0 | 0 | 1 | 0 |
| Abdominal pain | 1 | 0 | 0 | 0 |
| Nausea | 1 | 0 | 0 | 0 |
| Watery stool | 0 | 1 | 0 | 0 |
| Hyperglycemia | 1 | 0 | 0 | 0 |
| Dizziness | 5 | 10 | 3 | 1 |
| Faintness | 0 | 1 | 0 | 0 |
| Headache | 2 | 0 | 0 | 2 |
| Right leg cramps | 0 | 1 | 0 | 0 |
| Asymptomatic hypotension | 2 | 4 | 1 | 1 |
| Asymptomatic hypertension | 0 | 1 | 0 | 0 |
| Orthostatic hypotension | 4 | 3 | 0 | 0 |
| Symptomatic hypotension | 1 | 1 | 0 | 0 |
| Total adverse event | 19 | 26 | 6 | 4 |
Table 4: List of adverse events.
Discussion
A comparative randomized, open-label, single-dose, two-way crossover study, and with blinded determination of drug plasma concentrations was designed to comply with, ASEAN guideline for The Conduct of Bioequivalence Studies [15], European Medicines Agency (EMA) [16] and U.S. Food and Drug Administration (FDA) guidelines [17]. The bioequivalence studies of two different strengths of carvedilol tablets were conducted to compare the pharmacokinetic parameters of carvedilol between the test and reference products. Owing to the difference in composition between the formulations of test product of carvedilol 12.5 and 6.25 mg tablets, the bioequivalence study of carvedilol 6.25 mg tablets cannot be waived as per U.S. FDA product-specific guidance for generic drug development [18]. Thus, two separate bioequivalence studies were conducted for both strengths. Male and Female subjects were equally enrolled in the studies because the products are intended to be used in both genders. Sampling times were appropriately assigned since the mean % AUC extrapolation was less than 12% for both carvedilol and 4′-hydroxyphenyl carvedilol. This suggested that AUC0-t covered more than 80% of AUC0-∞ supporting the reliability of the concentration data, which accomplished the requirement of EMA guideline [16]. The 7-day washout period was sufficient for the elimination of carvedilol and its metabolite to be undetectable in plasma since the maximum half-lives of carvedilol and 4′-hydroxyphenyl carvedilol were around 11 and 15 hours, respectively and the assigned washout period was longer than 5 elimination half-lives as recommended by the EMA [16] and U.S. FDA guidelines [17].
The pharmacokinetics of carvedilol and 4′-hydroxyphenyl carvedilol conducted in healthy Thai subjects seem to be different compared to another bioequivalence study. In comparison, a bioequivalence study of carvedilol 12.5 mg tablets in healthy Indian male subjects under fed conditions showed lower AUC and Cmax than observed in the present study. This may cause from the reduced absorption by food as the study in Indian volunteers was conducted under fed states although food effect is not clinically relevant [19]. In addition, the gender difference may contribute to the increased mean AUC and Cmax values observed in the present study, in which female subjects were enrolled since the rate and extent of absorption of carvedilol have been reported to be higher in female subjects than in males [20].
From the ANOVA, there were no effects of formulation, period and sequence on the primary pharmacokinetic parameters of carvedilol and 4′-hydroxyphenyl carvedilol. The active metabolite of carvedilol, 4′-hydroxyphenyl carvedilol was also evaluated for its pharmacokinetics even though the data was not used for the bioequivalence evaluation. The 90% CIs for the ratios of geometric least square mean of ln-transformed primary pharmacokinetics parameter were well within the 80.00-125.00% range not only for carvedilol, but also the active metabolite 4′-hydroxyphenyl carvedilol in both bioequivalence studies. The bioequivalence of this active metabolite was supposed to be an occurrence from the same rate and extent of absorption of carvedilol of both formulations indicating that therapeutic equivalence could be anticipated. No serious adverse events occurred during conducting both studies indicating good tolerability of the formulations. The symptoms such as headache, dizziness and hypotension observed in the studies are commonly reported for the α- and β-adrenoreceptor blocking drugs [4]. In case of carvedilol, the vasodilatory effect resulted from R (+)-carvedilol contributing to α1-adrenoreceptor blockade seems to be a major cause of these symptoms, since R enantiomer is less metabolised by cytochrome P450 and substantially presented in systemic circulation [21]. The data from both bioequivalence studies were used for registration of generic carvedilol product, Carvolol 12.5 and 6.25 mg to Food and Drug Administration, Ministry of Public Health, Thailand.
Conclusion
The design of the study was suitable for the bioequivalence evaluation of carvedilol. Based on statistical indices, the test products, Carvolol 12.5 and 6.25 mg had the same rate and extent of absorption as the reference products, Dilatrend. The pharmacokinetics of carvedilol at the studies doses were dose proportional. The statistical comparison of active metabolite, 4′-hydroxyphenyl carvedilol illustrated no significant difference between the test and reference formulation, thus therapeutic equivalence can also be anticipated. Generally, the test and reference products were well tolerated by study subjects. The bioequivalence studies supported the use of generic products of carvedilol as alternatives to the innovator products which would enhance the accessibility to continued-use medication for patients at a reasonable price.
References
-
Sakboonyarat B, Rangsin R, Kantiwong A, Mungthin M (2019) Prevalence and associated factors of uncontrolled hypertension among hypertensive patients: A nation- wide survey in Thailand. BMC Res Notes 12(1): 1-8.
-
Farzam K, Jan A (2021) Beta blockers, StatPearls Publishing, Treasure Island, Florida, US.
-
Mctavish D, Campoli-Richards D, Sorkin EM (1993) Carvedilol a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 45(2): 232-258.
-
Keating GM, Jarvis B (2003) Carvedilol a review of its use in chronic heart failure. Drugs 63(16): 1697-1741.
-
Oldham HG, Clarke SE (1997) In vitro identification of the human cytochrome P450 enzymes involved in the metabolism of R(+)- and S(-)-carvedilol. Drug Metab Dispos 25(8): 970-977.
-
Zhou H-H, Wood AJJ (1995) Stereoselective disposition of carvedilol is determined by CYP2D6. Clin Pharmacol 57(5): 518-524.
-
Neugebauer G, Akpan W, Kaufmann B, Reiff K (1990) Stereoselective disposition of carvedilol in man after intravenous and oral administration of the racemic compound. Eur J Clin Pharmacol 38: S108-S111.
-
Portolés A, Filipe A, Almeida S, Terleira A, Vallée F, et al. (2005) Bioequivalence study of two different tablet formulations of carvedilol in healthy volunteers. Arzneimittelforschung 55(4): 212-217.
-
Fisker FY, Grimm D, Wehland M (2015) Third-generation beta-adrenoceptor antagonists in the treatment of hypertension and heart failure. Basic Clin Pharmacol 117(1): 5-14.
-
IPCA Laboratories Limited and India (2004) A randomized, open label, two treatments, two period, two sequence, single dose, cross over, bioequivalence study of carvedilol 25 mg tablets manufactured. India and Eucadic®25 mg tablet manufactured by Roach Products Limited, Welwyn Garden City, England.
-
World Medical Association Declaration of Helsinki (WMA) (2013) Ethical principles for medical research involving human subjects, 64th WMA General Assembly, Fortaleza, Brazil 310(20): 2191-2194.
-
WHO (2016) European Medicines Agency and International Conference on Harmonisation. Guideline for good clinical practice, ICH topic E6 (R2), London, United Kingdom.
-
EMA (2011) Guideline on bioanalytical method validation. Committee for Medicinal Products for Human Use (CHMP), London, United Kingdom.
-
Food and Drug Administration (2001) Guidance for industry: Bioanalytical method validation. Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM), Silver Spring, Maryland, US, pp: 28526-28527.
-
Food and Drug Administration and Ministry of Health (2015) ASEAN guideline for the conduct of bioequivalence studies, Vientiane, Lao PDR, Thailand.
-
EMA (2010) Guidance on the investigation of bioequivalence, CPMP/EWP/QWP/1401/98 Rev. 1, London, United Kingdom,pp: 1-27.
-
FDA (2013) Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA. Center for Drug Evaluation and Research (CDER), Silver Spring, Maryland, US, pp:1-38.
-
FDA and U.S. Department of Health and Human Services (2008) Product-specific guidances for generic drug development: Guidance on carvedilol. Center for Drug Evaluation and Research (CDER), Silver Spring, Maryland, US.
-
Morgan T (1994) Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet 26(5): 335-346.
-
Abbas M, Muqeet Khan A, Sualeha R, Tipu Y, Awais Nawaz H, et al. (2014) Assessment of sex differences in pharmacokinetics of carvedilol in human. J Pharm Sci 27(5):1265-1269.
-
Taniguchi T, Ohtani T, Mizote I, Kanzaki M, Ichibori Y, et al. (2013) Switching from carvedilol to bisoprolol ameliorates adverse effects in heart failure patients with dizziness or hypotension. J Cardiol 61(6): 417-422.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review