Basic Aspects and the Overview on Pharmacokinetics and Studies on Drug Distribution- A Panoramic Review
Patients who are in critical condition display a variety of organ dysfunctions and frequently need to be treated with a range of medications, such as sedatives, analgesics, neuromuscular blockers, antibiotics, inotropes, and gastric acid suppressants. A crucial component of treatment for this patient population is comprehending how organ dysfunction might change the pharmacokinetics of medications. Due to gastrointestinal failure, many medications will need to be administered intravenously. When the oral route is an option, hypomotility, changes in gut pH, and enteral feeding may affect bioavailability. The main factors affecting medication clearance, and consequently steady-state drug concentrations, efficacy, and toxicity in a given patient, are hepatic and renal dysfunction. Many medications are cleared from the body primarily through oxidative metabolism, and it is becoming increasingly understood how important it is for critically ill patients to have diminished hepatic cytochrome P450 system activity. Both filtration and secretion clearance pathways are necessary for the elimination of parent medications and their active metabolites, making renal failure equally crucial. Renal failure is frequently a secondary cause of changes in the steady-state volume of distribution, which can lower the body's effective medication concentrations. Failure of the endocrine, endothelium, muscular, or central neurological systems may also have an impact on how a medication is metabolized. For some medications, there is strong evidence that changes in pharmacokinetic characteristics depend on time. To maximize the pharmacodynamic response and result, it is essential to understand the underlying pathophysiology in the critically sick and utilize pharmacokinetic principles in the selection of drug and dosing regimen.
Introduction
What is Biopharmaceutics?
In the world of drug development, the meaning of the term “Biopharmaceutics” often causes confusion, even among scientists and professionals working in the field. “Pharmaceuticals” in the narrow sense is a field of science that includes the manufacture, use, or pharmacy of pharmaceuticals. The addition of the prefix “bio”, derived from the Greek word “bios”, refers to an organism or tissue, expanding this field to include the manufacture, use, and administration of drugs into an organism or tissue include science [1, 2]. The concept of bio-pharmacy described here has an inherent interdependence between the biological aspects of the living body (patient) and the physicochemical principles that govern the manufacture and behavior of the drug. This philosophy was developed in the mid-20th century by the first generation of Biopharmaceutics scientists. He is now called a Biopharmaceutics scientist. Those who are aware of the importance of absorption, distribution, metabolism and excretion (ADME) in the clinical outcome of drugs say: Also, the effect of the physicochemical properties of the material on the performance in vivo [3]. As a result, biopharmaceuticals have evolved into a wide range of disciplines that incorporate basic principles in basic science and related disciplines, such as chemistry, physiology, physics, statistics, engineering, mathematics, microbiology, enzymology, and cell biology [4, 5].
Biopharmaceutics: Biopharmaceutics involves studying the physicochemical properties of a drug, the dosage form in which the drug is administered, and the interrelationships between routes of administration regarding the rate and extent of systemic drug absorption [1].
Absorption: The process of unchanged drug transfer from the site of administration to the systemic circulation [1].
Passive diffusion: It involves transport of the drug to a concentration gradient from a low drug concentration region to a high concentration until equilibrium [1].
Pharmacokinetics: Pharmacokinetics is defined as the kinetics of drug absorption, distribution, metabolism and excretion (ADME) and their relationship with the pharmacological, therapeutic or toxicological response in man and animals [1].
Pharmacokinetics
Pharmacokinetics has been proposed to study drug absorption, distribution, in vivo changes, and excretion in humans and animals. A more complete description of the data can be obtained by interpolating and extrapolating the drug concentration using several mathematical functions [6]. You can use these functions to reduce all the data to a smaller set of parameters, or to see if the hypotheses contained in the function are supported by the observations. In the first case, the task is to get a simulation of the data, and in the second case, to get a model. The function used to interpolate and reduce pharmacokinetic data is a multi-exponential function, the reference model is a compartment model, and its solution is only a multi-exponential function [7, 8]. Models can be used to define new and meaningful pharmacokinetic parameters that can be used to find relationships between drug dynamic profiles and physiological processes that promote drug absorption, distribution, and excretion as per shown in Figure 1. For example, a compartment model makes it easy to define a clearance that depends on the drug removal process, or a volume of distribution that depends on the drug distribution in the tissue [9]. The model also provides an easy way to obtain an estimate of drug absorption (bioavailability) after extravascular drug administration [10]. Modeling is a complex, multi-step process in which new hypotheses are proved and disproved by experimental and simulated simulations through continuous interaction between the experimenter and the computer.
![Figure 1: For example, a compartment model makes it easy to define a clearance that depends on the drug removal process, or a volume of distribution that depends on the drug distribution in the tissue [9]. The model also provides an easy way to obtain an estimate of drug absorption (bioavailability) after extravascular drug administration [10]. Modeling is a complex, multi-step process in which new hypotheses are proved and disproved by experimental and simulated simulations through continuous interaction between the experimenter and the computer.](/fulltextimages/9633/fig_1.png)
Absorption
Mechanism of drug absorption the main mechanisms by which absorption occurs include:
- Transcellular/Intracellular Transport is defined as the passage of drugs across the GI epithelium. It is the most common pathway for drug transport.
- Paracellular/Intercellular Transport – is defined as the transport of drugs through the junctions between the GI epithelial cells. This pathway is of minor importance in drug absorption.
- Vesicular or Corpuscular Transport (Endocytosis) – Like active transport, these are also energy dependent processes but involve transport of substances within vesicles into a cell. Since the mechanism involves transport across the cell membrane, the process can also be classified as Transcellular [11, 12, 13].
Mechanism of drug absorption • Passive Diffusion: A lipophilic medication may bypass or enter the cell. Medication diffusion and absorption are not impeded by the lipid cell membrane if the drug is lipophilic and has a low molecular weight. Molecules will naturally move from a location of higher concentration to a region of lower concentration through a process called passive diffusion. Since no outside energy is used, this process is passive [14, 15, 16].
• Carrier-Mediated Transport: A lipophilic medication might theoretically bypass or enter the cell. Medication diffusion and absorption are not impeded by the lipid cell membrane if the drug is lipophilic and has a low molecular weight. Both diffusion and a carrier-mediated method can be used in the intestine to pass medicines and other substances through the intestinal epithelial cells. The body has several different specialized carrier- mediated transport systems, many of which are located in the intestine where the body absorbs ions and nutrients [14, 15, 16].
• Active Transport: Active transport is a carrier-mediated transmembrane process that is crucial for the renal and biliary secretion of numerous medicines and metabolites as well as the absorption of these substances through the gastrointestinal tract. This method allows for the absorption of a few lipid-insoluble medications (such as 5-fluorouracil), which resemble natural physiologic metabolites. Drugs are transported actively when they move against a concentration gradient, or when they move from areas with low drug concentrations to areas with high concentrations. As a result, this system uses a lot of energy. Additionally, active transport is a specialized procedure that calls for a carrier that binds the drug to create a carrier-drug complex that transports the drug across the membrane before dissociating it on the other side [14, 15, 16].
• Vesicular Transport: The movement of a protein, like insulin, out of the cells of the pancreas into the extracellular space is an example of exocytosis. In order to release the insulin outside the cell, the insulin molecules are first bundled into intracellular vesicles, which subsequently fuse with the plasma membrane [14, 15, 16].
• Pore (Convective) Transport: Very small molecules can quickly traverse cell membranes (including urea, water, and carbohydrates), as if the membrane had channels or pores. The hypothesis of drug penetration through aqueous pores is employed to explain renal excretion of medications and the absorption of drugs into the liver, despite the fact that such pores have never been directly detected by microscopy [14, 15, 16].
• Ion-Pair Formation: Strong electrolyte medications are molecules that are highly ionized or charged, including quaternary nitrogen compounds with extremely high pKa values. Drugs that contain strong electrolytes have trouble penetrating membranes and retain their charge at all physiological pH levels. An ion pair with a neutral overall charge is created when the ionized medication is coupled with an oppositely charged ion. This medication combination diffuses more freely through the membrane because it is neutral. Propranolol, a basic medication that forms an ion pair with oleic acid, and quinine, which forms an ion pair with hexylsalicylate, is two examples of how the formation of ion pairs can help in drug absorption has been established [14, 15, 16].
Drug Distribution
Drug distribution was referring to the reversible transfer of a drug between the blood and the extravascular fluids and tissues of the body. Drugs come into the circulation after absorption. From plasma, drugs have to cross the capillary membrane to come to interstitial space and then need cross the cell-membrane to enter into the intracellular fluid [17].
Factors Affecting Distribution of Drugs
- Tissue Permeability of the Drug [18] a) Physiochemical Properties of the drug like Molecular size, pKa and o/w Partition coefficient. b) Physiological Barriers to Diffusion of Drugs.
- Organ / Tissue Size and Perfusion Rate [19]
- Binding of Drugs to Tissue Components [20] a) Binding of drug to blood components b) Binding of drug to extra cellular components c) Miscellaneous Factors [21] Age, Pregnancy, Obesity, Diet, Disease states, and Drug Interactions etc.
- Tissue Permeability of the Drugs depends upon [22]: a) Rate of Tissue Permeability, and b) Rate of Blood Perfusion. The Rate of Tissue Permeability depends upon Physiochemical Properties of the drug as well as Physiological Barriers that restrict the diffusion of drug into tissues.
Drug Metabolism
It is the conversion of a drug molecule into other related compounds throughout the body. The modification of an administered drug is a normal process carried out by drug modifying enzymes (DME) such as cytochrome P450 [23]. Even though drug metabolism in numerous locations within the body, the liver is the primary organ involved in the modification and removal of drug compounds. Metabolism qualities of a drug candidate encompass any modification to it by bodily organs or enzymes [24]. This process is one of the most important parts of the DMPK process since these data outputs are closely linked to the potential efficacy, or toxicity, of a drug when it enters the human body.
In actuality, the body seeks to eliminate or detoxify any material that cannot be used in another way to meet its demands [25]. That is the essence of what metabolism implies. Altering a xenobiotic to facilitate elimination from the body. The liver is primarily responsible for carrying out this elimination procedure. Numerous enzymes found in the liver are quite good at doing this job. Two phases of drug metabolism can be distinguished. Phase 1 metabolism is made up of processes including oxidation, reduction, and hydrolysis and is mostly carried out by cytochrome P450s (CYPs) and monooxygenases that include flavin [26]. These phase 1 reactions serve two purposes: either to prepare for phase 2 reactions that add a water-soluble endogenous molecule to lower lipophilicity or to reduce the lipophilicity sufficiently to promote renal clearance [27].
CYPs are among the most researched and thoroughly characterised enzyme groups because they are the most significant enzymes in drug metabolism. At every stage of the drug development process, these enzymes are investigated. One of the first tests used in DMPK is presently screening libraries of chemicals for CYP inhibition [28]. A CYP inhibitor can change physiological processes by interfering with regular cellular metabolism. If the same enzyme is involved in the metabolism of two different medications, patients who are currently on one are sometimes recommended against or banned from taking another medicine [29, 30]. As a result, there may be what are known as drug-drug interactions, in which the slower metabolism of one medicine results in an undesirable side effect.
It is possible to forecast in vivo metabolism using in vitro metabolism. The DMPK scientist can leverage the capacity to separate cellular fractions equipped with knowledge of those fractions’ characteristics, whether they be microsomal, whole cell, or even recombinant enzymes. Robust assays are used throughout the industry [31]. The in vitro metabolic pathway can be clarified with information of the enzymatic make-up of the specific cellular fraction because each fraction will display various cell components. Calculating intrinsic clearance is an objective of in vitro work with cellular fractions. This is frequently carried out in a number of species, and the data is then extrapolated to produce a forecast of human clearance.
Excretion of Drugs
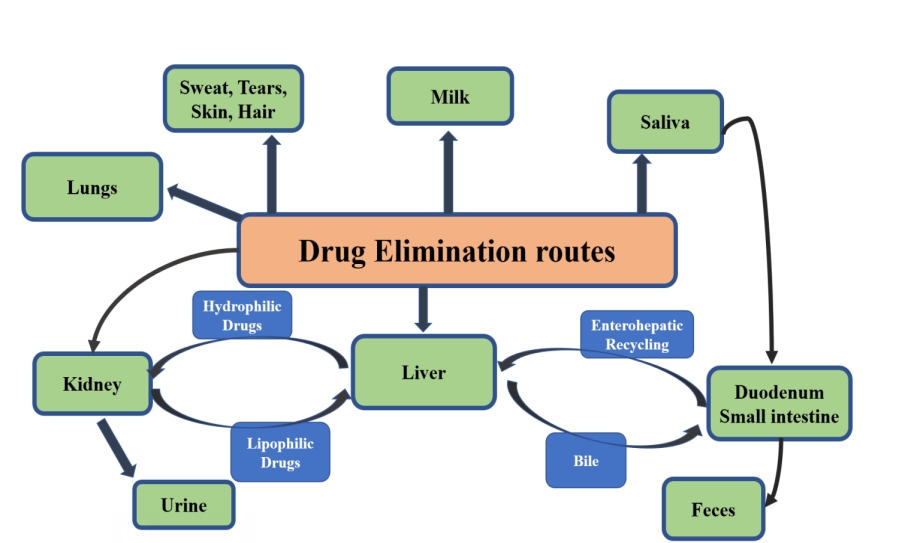
Excretion is defined as the process whereby drugs and/ or their metabolites are irreversibly transferred from internal to external environment. Kidney is the most important organ involved in excretion of most drugs especially which are water soluble or having low molecular weight. Nephron is the functional unit of kidney consisting of various parts viz glomerulus, proximal tubule, loop of Henle, distal tubule and the collecting tubule [32].
Some medications and drug metabolites are removed from the body by biliary excretion in humans. The features of the medication, such as chemical structure, polarity, and molecular size, as well as the characteristics of the liver, such as specific active transport sites within the liver cell membranes, determine clearance via the biliary route as per shown in Figure 2 [33]. A drug that has been expelled in bile may be reabsorbed from the digestive system, or a drug conjugate may be hydrolyzed by gut bacteria, releasing the original drug that may then be reintroduced to the bloodstream [33, 34, 35]. The pharmacological impact of some medications and drug metabolites may be prolonged through enterohepatic circulation, but the quantitative significance of this in humans appears to be smaller than in animals.
The interindividual variations in medication response seen in both healthy persons and patients with specific illnesses may be influenced by biliary clearance. Drug clearance via this route will be influenced by cholestatic illness conditions, in which normal bile flow is impaired, increasing the risk of drug toxicity [36]. Bile may act as a secondary elimination pathway in renal failure, however there is no evidence for this in humans. The relative inaccessibility of the human biliary tract is one reason why there is a lack of trustworthy information regarding the biliary excretion of pharmaceuticals in man. The majority of research on medication excretion in human bile has been done on post-operative patients receiving T-tube drainage [37]. This method of bile collection is not optimal since enterohepatic circulation is partially disrupted, bile flow and composition are frequently significantly changed throughout the study period, and some bile is not collected [38]. Future research on medication excretion in human bile may benefit from recent improvements in bile collection techniques.
Renal excretion refers to the process by which drugs are excreted through kidney. The major excretory processes that determine the excretion are [39]: I. Glomerular filtration 2 Active tubular secretions 3. Tubular reabsorption
Distribution of Drugs
Drug delivery is represented by volume of distribution, one of the key pharmacokinetic parameters. To address the importance of the pharmacokinetic of the distribution, its conceptual relationship with other pharmacokinetic parameters is important, including clearance as the primary parameter and the rejection rate constant as the parameter extra [13]. In addition, different mass distribution parameters exist, which are inferred from different models and analytical approaches. A simple explanation to describe these distribution parameters is helpful. Importantly, the volume of distribution is significantly altered, as is the clearance, during the physiology of pregnancy and during growth and development [40]. Pregnancy is characterized by fluid dilation and increased perfusion of the excretory organs, causing an increase in volume of distribution and clearance with few exceptions. Distribution of drugs in breast milk is often discussed with placental transmission, but their pharmacokinetic mechanisms are very different. Infants and children also have increased volume of distribution and increased body weight clearance [41, 42, 43, 44, 45]. However, their development configuration is sensitive to unit standardization. Although the volume of distribution is linearly consistent with body weight after infancy, the clearance is not, but is proportional to the corresponding capacity model of (Weight) 0.75, which is can be explained by the correlation relationship between liver/kidney growth and body mass gain.
Distributing Medicine to Children
Drug distribution affects efficacy and duration of action. Ginsberg, et al. Compared the pharmacokinetic parameters of 45 drugs in children and adults and concluded that there is a trend toward a greater mass distribution of these compounds in children of all age groups [46]. Distribution of the drug depends on body composition. Lipidants have a relatively larger volume of distribution in neonates than in older children due to their higher fat content (22.4% in 12-month infants vs. 13% at age 15-32). For example, diazepam, a lipophilic drug, has a volume of distribution in adults and a ratio in neonates of 0.7 [47, 48]. Hydrophilic drugs also have a greater volume of distribution in preschool children, as extracellular fluid decreases during development, from 70% of total body weight in neonates to 61.2% in infants [49]. One-year-old infant. Because of the higher volume of distribution of water-soluble drugs in neonates, e.g. gentamicin, higher doses per kg body weight should be given to neonates than to adults to achieve plasma and tissue concentrations equivalent [50]. Protein binding also affects the volume of distribution of the drug. In neonates, the total plasma protein concentration is 86% of the adult value. In general, it can be assumed that the effect of protein binding on plasma free drug concentrations is limited to drugs with a high degree of protein binding (> 95%). When protein levels reach adult values during the neonatal period, this effect may be more pronounced in neonates and infants.
Distribution of Drugs in Adults
A drug will deliver to different tissues or organs after reaching the circulatory system. Distribution morphology will depend in part on physical (e.g., lipophilic or water solubility, degree of ionization) and physiological (e.g., protein binding, tissue absorption). Distribution is therefore dependent on protein binding, pH, systemic and regional blood flow, permeability of natural “barriers” (e.g., blood-brain, placenta), and body part [51]. Obviously, these covariates would display both inter- and intra-patient variability, partly explained by changes in adulthood or disease-related differences. The effect of body structure mature age-dependent changes in body composition alter the physiological space into which the drug will distribute [52]. Newborns and infants have a proportionally higher amount of body water per kilogram of body weight than children and adults, and premature infants have this value even higher than infant’s full-term birth. As for the total body water content, this is about 80% to 90% in premature infants and 70% in full-term infants, gradually decreasing to about 60% by the end of the first 1-2 years and after that is stable throughout childhood [53]. The pattern is similar for extracellular water content, which begins at 40% and declines to about 25% to 30% by late infancy. For lipid compartments, the trend is somewhat more complicated, with an initial increase from 10% to 15% at birth to 20% to 25% in late infancy, and then a decrease to 10% to 15% until adolescence. The greater total body space and foreign body space in neonates and neonate’s results in lower plasma drug concentrations for these respective compartmentalized drugs when administered by weight [54]. The opposite is true for lipophilic compounds. To illustrate this, the volume of distribution of (water-soluble) aminoglycosides shows a gradual decrease in the neonatal period (very preterm, 0.7 L/kg, at term, 0.5 L/kg) and throughout childhood (0.5 L/kg in infants. to 0.3 L/kg in young adults). Similar patterns have been described for paracetamol (acetaminophen). In contrast, diazepam, a lipophilic compound, had a correspondingly lower volume of distribution in neonates (1.6 L/kg) than in children or adults (2.4 L/kg). One Similar trend was estimated for protocol, another lipophilic compound (2.8- 5.6 L/kg in infants to 5.6-8.6 L/kg in toddlers) throughout the neonatal period. Protein binding Protein binding also affects drug delivery [55]. Compared with adults, infants and children have lower concentrations of plasma best- binding proteins such as albumin, acid α-1 glycoprotein or plasma globulins. Since protein concentrations reach adult values during the neonatal period, this effect may be more pronounced in infants and young children [56]. In addition to absolute values or concentrations, competitive binding with endogenous compounds (e.g., bilirubin, free fatty acids) may further affect binding capacity.
In neonates, there is concomitantly lower concentrations of various plasma proteins associated with increased concentrations of bilirubin and/or increased concentrations of free fatty acids. In addition, the amount and type of circulating plasma proteins will affect not only the disposition of the drug but also its activity, since only unbound or free drug can be distributed throughout the body [57]. And exert pharmacological effects. The clinical significance of altering drug protein binding is most relevant to drugs with high protein binding and also with a narrow therapeutic index. A recently published example to illustrate this is the protein binding of cefazolin to albumin: the free fraction is related to the total concentration of cefazolin as well as to the amount of serum albumin, but even after combining covariates in this case, the free fraction is still higher in infants [58]. Similar trends have been described for other antibiotics (e.g., ampicillin, flucloxacillin, vancomycin), antiepileptic drugs (e.g., phenytoin), or chemotherapeutic agents (e.g., etoposide). The reduction in protein binding increases the free concentration and free fraction of the drug, thereby enhancing the easier diffusion of the active substance to other compartments [59]. This will cause a greater interaction with the receptors, but it will also increase the CL concentration of the drug. In addition, when higher free fractions of a given compound circulate in the plasma compartment, these fractions can enter deeper tissue compartments, resulting in a higher volume of distribution. These principles must be borne in mind when prescribing medications to these patients [60]. For drugs with a defined therapeutic serum concentration relationship (e.g. theophylline), target concentrations will be lower, exhibit less protein binding/higher free drug concentrations in neonates. Birth (e.g., apnea in preterm infants) compared with target concentrations in older infants, children, and adults (e.g., asthma). In summary, the effect of protein binding on plasma free drug concentrations is limited to drugs with moderate to high degree of protein binding and a narrow therapeutic index (e.g., phenytoin, etoposide) [61]. Indeed, even a small difference in protein binding will result in a significant difference in the free concentration of the drug used. Membrane permeability Drug delivery to deep compartments such as the central nervous system is slowed and limited due to tight junctions of the endothelium associated with flow transporters [62]. This is why intracavity injections are performed to overcome this barrier in children, for example, with acute leukemia or brain tumors. Although the number of observations is still limited, it appears that there are also mature changes in this barrier, with progressive increases in flow transporter (P- gp) expression and function as well [63]. As tight junction (passive diffusion is higher at infancy) capacitance.
Distributional Differences Caused by Disease
Estimates of the volume of phenotypic distribution throughout childhood are to a large extent determined by changes during adulthood. However, these estimates may be further influenced by disease-related changes in body composition, protein binding capacity, or membrane permeability [64]. Obesity and malnutrition are significant in children. Currently, there is no universal descriptor to estimate the volume of distribution of all drugs in lean and obese children, but it can be predicted that higher fat mass primarily alters the distribution of drugs of lipophilic drugs and have more limited effects on water-soluble compounds [65, 66, 67, 68]. Based on the available evidence, protein malnutrition does not significantly affect the mass distribution of most compounds, but affects absorption and CL to a greater extent. Similarly, the presence of persistent dictums arteriosus or sepsis was associated with a further increase in the volume of distribution in term (pre)term neonates, which is more evident for soluble compounds country [69, 70]. In addition, the use of oxygen across the extracorporeal membrane will affect drug distribution because of the additional external volume (membrane and tubules) as well as the low plasma protein content and associated fluid retention commonly seen. Protein binding is clearly affected in hypoalbuminemia (e.g., nephrotic syndrome), but environmental aspects (pH, free fatty acids, competitive binding) may also influence association characteristics Protein binding [71]. Competitive binding is a major problem in neonates with hyperbilirubinemia because displacement of the initially bound bilirubin can lead to kernicterus [72, 73]. Therefore, ceftriaxone should not be given to infants with high bilirubin levels. Binding characteristics may also be related to disease status, as the acid α-1 glycoprotein is increased postoperatively, resulting in a slightly greater binding capacity for local anesthetics [74]. Finally, in addition to the maturation changes, membrane permeability can also be affected by the disease state. In the setting of meningitis, the associated inflammation would lead to less efficient tight junctions, and a similar pattern can be predicted in the case of (severe) traumatic brain injury [74].
Conclusion
A chemical must be able to reach its targeted receptor sites in vivo in sufficient quantity and for the necessary amount of time to exert a biological impact, in addition to possessing selective effectiveness against the molecular target(s). After being administered in vivo, a substance’s fate is determined by its target residence duration, as well as its absorption, distribution, metabolism, and excretion (ADME). The culmination of all of these activities is represented by the compound’s concentration in the blood, plasma, and other tissues. The procedures for giving a substance to rats via various routes and for gathering the necessary samples to ascertain the pharmacokinetics profile are covered in this unit.
Although there is conclusive information on how ageing affects an animal’s ability to metabolize drugs, no direct evaluations have been done in humans. Changes in distribution challenge a lot of the research that use medication plasma half-life and clearance estimations. The best example of this comes from a conclusive study with diazepam, where a clearly extended plasma half-life was followed by an enhancement in apparent volume of distribution in aged patients. The latter modification affects drug concentration at the site of action as well as plasma drug clearance. Therefore, it is yet unclear what these changes in distribution volume mean for the effects of drugs.
Declarations
Consent for publication
Nil
Availability of Data and Material
Not Applicatble
Authors’ Contributions
All the authors have contributed to the research work and preparation of the final manuscript.
Conflict Of Interests
The authors declare no conflict of interests.
Acknowledgments
No acknowledgments
Ethical Declaration
No animal or human subjects were used during the preparation of this manuscript.
Funding
No
References
-
Yeh MK, Chen YC (2018) Biopharmaceuticals: BoD– Books on Demand.
-
Price G, Patel DA (2022) Drug bioavailability. StatPearls Publishing, Treasure Island (FL).
-
Kesik-Brodacka M (2018) Progress in biopharmaceutical development. Biotechnol Appl Biochem 65(3): 306-322.
-
Eibl D, Eibl R (2019) Single‐use equipment in biopharmaceutical manufacture: a brief introduction. 2nd(Edn.), John Wiley & Sons, Inc, pp: 1-11.
-
Rosales-Mendoza S, Solís-Andrade KI, Márquez- Escobar VA, González-Ortega O, Bañuelos-Hernandez B, et al. (2020) Current advances in the algae-made biopharmaceuticals field. Expert Opin Biol Ther 20(7): 751-766.
-
Zhao M, Wang Y, Chen M, Wu B (2020) Introduction to Pharmacokinetics. Circadian Pharmacokinetics, Springer, pp: 23-40.
-
Currie GM (2018) Pharmacology, part 2: introduction to pharmacokinetics. J Nucl Med Technol 46(3): 221-230.
-
Guidi M, Csajka C, Buclin T (2022) Parametric approaches in population pharmacokinetics. J Clin Pharmacol 62(2): 125-141.
-
Kabadi SV, Lin Z (2020) Introduction to classical pharmacokinetics. Physiologically Based Pharmacokinetic (PBPK) Modeling, pp: 27-56.
-
Talevi A, Quiroga PA (2018) Introduction. Biopharmaceutics and pharmacokinetics. ADME Processes in Pharmaceutical Sciences, Springer, pp: 3-10.
-
Vinarov Z, Abdallah M, Agundez JAG, Allegaert K, Basit AW, et al. (2021) Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur J Pharm Sci 162: 105812.
-
Barros AS, Costa A, Sarmento B (2021) Building three- dimensional lung models for studying pharmacokinetics of inhaled drugs. Adv Drug Deliv Rev 170: 386-395.
-
van den Anker J, Reed MD, Allegaert K, Kearns GL (2018) Developmental changes in pharmacokinetics and pharmacodynamics. J Clin Pharmacol 58(10): 10-25.
-
Di L, Artursson P, Avdeef A, Benet LZ, Houston JB, et al. (2020) The critical role of passive permeability in designing successful drugs. ChemMedChem 15(20): 1862-1874.
-
Alagga AA, Gupta V (2022) Drug absorption. StatPearls Publishing, Treasure Island (FL).
-
Rubbens J, Mols R, Brouwers J, Augustijns P (2018) Exploring gastric drug absorption in fasted and fed state rats. Int J Pharm 548(1): 636-641.
-
Tetro N, Moushaev S, Rubinchik-Stern M, Eyal S (2018) The placental barrier: the gate and the fate in drug distribution. Pharm Res 35(4): 71.
-
Olusanya TOB, Haj Ahmad RR, Ibegbu DM, Smith JR, Elkordy AA, et al. (2018) Liposomal drug delivery systems and anticancer drugs. Molecules 23(4): 907.
-
Khosa A, Reddi S, Saha RN (2018) Nanostructured lipid carriers for site-specific drug delivery. Biomed Pharmacother 103: 598-613.
-
Ding Y, Li W, Zhang F, Liu Z, Zanjanizadeh Ezazi N, et al. (2019) Electrospun fibrous architectures for drug delivery, tissue engineering and cancer therapy. Advanced Functional Materials 29(2): 1802852.
-
Bhaskar M, Telessy IG, Buttar HS (2022) The Importance of Drug Dose Adjustment in Elderly Patients with Special Considerations for Patients on Diverse Co-medications and Antidepressants. Biomedical Translational Research, pp: 231-272.
-
Boyd BJ, Bergström CAS, Vinarov Z, Kuentz M, Brouwers J, et al. (2019) Successful oral delivery of poorly water- soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur J Pharm Sci 137: 104967.
-
Zhang Z, Tang W (2018) Drug metabolism in drug discovery and development. Acta Pharm Sin B 8(5): 721- 732.
-
Li Y, Meng Q, Yang M, Liu D, Hou X, et al. (2019) Current trends in drug metabolism and pharmacokinetics. Acta Pharm Sin B 9(6): 1113-1144.
-
He C, Wan H (2018) Toxicology. Drug metabolism and metabolite safety assessment in drug discovery and development. Expert Opin Drug Metab Toxicol 14(10): 1071-1085.
-
de Jong LM, Jiskoot W, Swen JJ, Manson ML (2020) Distinct effects of inflammation on cytochrome P450 regulation and drug metabolism: lessons from experimental models and a potential role for pharmacogenetics. Genes (Basel) 11(12): 1509.
-
Zhao M, Ma J, Li M, Zhang Y, Jiang B, et al. (2021) Cytochrome P450 enzymes and drug metabolism in humans. Int J Mol Sci 22(23): 12808.
-
Doohan PT, Oldfield LD, Arnold JC, Anderson LL (2021) Cannabinoid interactions with cytochrome P450 drug metabolism: a full-spectrum characterization. AAPS J 23(4): 91.
-
Pike A, Williamson B, Harlfinger S, Martin S, McGinnity DF, et al. (2020) Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today 25(10): 1793-1800.
-
Serras AS, Rodrigues JS, Cipriano M, Rodrigues AV, Oliveira NG, et al. (2021) A critical perspective on 3D liver models for drug metabolism and toxicology studies. Front Cell Dev Biol 9: 626805.
-
Ooka M, Lynch C, Xia M (2020) Application of in vitro metabolism activation in high-throughput screening. Int J Mol Sci 21(21): 8182.
-
Knott C (2021) Excretion of drugs into saliva. In: Tenovuo JO, et al. (Eds.), Human saliva: clinical chemistry and microbiology. CRC Press, pp: 264.
-
Kundu P, Das S, Chattopadhyay N (2019) Managing efficacy and toxicity of drugs: Targeted delivery and excretion. Int J Pharm 565: 378-390.
-
Xiao J, Tran D, Zhang X, Zhang T, Seo S, et al. (2020) Biliary Excretion–Mediated Food Effects and Prediction. AAPS J 22(6): 124.
-
Anderson PO (2018) Drugs in lactation. Pharm Res 35(3): 45.
-
Smith DA, Beaumont K, Maurer TS, Di L (2018) Clearance in drug design: miniperspective. J Med Chem 62(5): 2245-2255.
-
Jansen K, Pou Casellas C, Groenink L, Wever KE, Masereeuw R, et al. (2020) Humans are animals, but are animals human enough? A systematic review and meta-analysis on interspecies differences in renal drug clearance. Drug Discov Today 25(4): 706-717.
-
Blanco VE, Hernandorena CV, Scibona P, Belloso W, Musso CG, et al. (2019) Acute kidney injury pharmacokinetic changes and its impact on drug prescription. Healthcare (Basel) 7(1): 10.
-
Andrikou C, Thiel D, Ruiz-Santiesteban JA, Hejnol A (2019) Active mode of excretion across digestive tissues predates the origin of excretory organs. PLoS Biol 17(7): e3000408.
-
Shultz MD (2018) Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J Med Chem 62(4): 1701-1714.
-
Ke AB, Greupink R, Abduljalil K (2018) Drug dosing in pregnant women: challenges and opportunities in using physiologically based pharmacokinetic modeling and simulations. CPT Pharmacometrics Syst Pharmacol 7(2): 103-110.
-
Koren G, Pariente G (2018) Pregnancy-associated changes in pharmacokinetics and their clinical implications. Pharm Res 35(3): 61.
-
Dallmann A, Mian P, Van den Anker J, Allegaert K (2019) Clinical pharmacokinetic studies in pregnant women and the relevance of pharmacometric tools. Curr Pharm Des 25(5): 483-495.
-
Kazma JM, van den Anker J, Allegaert K, Dallmann A, Ahmadzia HK, et al. (2020) Anatomical and physiological alterations of pregnancy. J Pharmacokinet Pharmacodyn 47(4): 271-285.
-
Dallmann A, Pfister M, van den Anker J, Eissing T (2018) Physiologically based pharmacokinetic modeling in pregnancy: a systematic review of published models. Clin Pharmacol Ther 104(6): 1110-1124.
-
Lee JH, Byon HJ, Choi S, Jang YE, Kim EH, et al. (2018) Safety and efficacy of off-label and unlicensed medicines in children. J Korean Med Sci 33(37): e227.
-
Theusinger OM, Schenk P, Dette-Oltmann K, Mariotti S, Baulig W, et al. (2019) Treatment of seizures in children and adults in the emergency medical system of the city of Zurich, Switzerland–midazolam vs. diazepam–a retrospective analysis. J Emerg Med 57(3): 345-353.
-
Mostafavi SN, Jafari A, Hoseini SG, Khademian M, Kelishadi R, et al. (2019) The efficacy of low and moderate dosage of diazepam on sleep bruxism in children: A randomized placebo-controlled clinical trial. J Res Med Sci 24: 8.
-
Zhang Y, Shen L, Wang T, Li H, Huang R, et al. (2020) Taste masking of water-soluble drug by solid lipid microspheres: A child-friendly system established by reversed lipid-based nanoparticle technique. J Pharm Pharmacol 72(6): 776-786.
-
Shahrin L, Chisti MJ, Sarmin M, Rahman ASMMH, Shahid ASMSB, et al. (2021) Intravenous amoxicillin plus intravenous gentamicin for children with severe pneumonia in Bangladesh: An open-label, randomized, non-inferiority controlled trial. Life (Basel) 11(12): 1299.
-
Thürmann PA (2020) Pharmacodynamics and pharmacokinetics in older adults. Curr Opin Anaesthesiol 33(1): 109-113.
-
Goldman JE, Waye KM, Periera KA, Krieger MS, Yedinak JL, et al. (2019) Perspectives on rapid fentanyl test strips as a harm reduction practice among young adults who use drugs: a qualitative study. Harm Reduct J 16(1): 3.
-
Rajan J, Behrends M (2019) Acute pain in older adults: recommendations for assessment and treatment. Anesthesiol Clin 37(3): 507-520.
-
Mehrotra S, Bhattaram A, Krudys K, Bewernitz M, Uppoor R, et al. (2022) Extrapolation of Efficacy from Adults to Pediatric Patients of Drugs for Treatment of Partial Onset Seizures (POS): A Regulatory Perspective. Clin Pharmacol Ther 112(4): 853-863.
-
Hahn RG (2020) Understanding volume kinetics. Acta Anaesthesiol Scand 64(5): 570-578.
-
Tu Y, Yu Y, Zhou Z, Xie S, Yao B, et al. (2019) Specific and quantitative detection of albumin in biological fluids by tetrazolate-functionalized water-soluble AIEgens. ACS Appl Mater Interfaces 11(33): 29619-29629.
-
Hill MD, Abramson FP (1988) The significance of plasma protein binding on the fetal/maternal distribution of drugs at steady-state. Clin Pharmacokinet 14(3): 156- 170.
-
Xie V (2022) Understanding drug–protein binding and ADME studies for DMPK. Bioanalysis 14(13): 919-921.
-
Bennett CF, Kordasiewicz HB, Cleveland DW (2021) Antisense drugs make sense for neurological diseases. Annu Rev Pharmacol Toxicol 61: 831-852.
-
Carles F, Bourg S, Meyer C, Bonnet P (2018) PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 23(4): 908.
-
Koziolek M, Alcaro S, Augustijns P, Basit AW, Grimm M, et al. (2019) The mechanisms of pharmacokinetic food- drug interactions–A perspective from the UNGAP group. Eur J Pharm Sci 134: 31-59.
-
Wanat K (2020) Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol Biol Rep 47(4): 3221-3231.
-
Bakos É, Német O, Patik I, Kucsma N, Várady G, et al. (2020) A novel fluorescence‐based functional assay for human OATP1A2 and OATP1C1 identifies interaction between third‐generation P‐gp inhibitors and OATP1A2. FEBS J 287(12): 2468-2485.
-
Su C, Liu Y, Li R, Wu W, Fawcett JP, et al. (2019) Absorption, distribution, metabolism and excretion of the biomaterials used in Nanocarrier drug delivery systems. Adv Drug Deliv Rev 143: 97-114.
-
Zucker I, Prendergast BJ (2020) Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol Sex Differ 11(1): 32.
-
Padrini R (2019) Clinical pharmacokinetics and pharmacodynamics of direct oral anticoagulants in patients with renal failure. Eur J Drug Metab Pharmacokinet 44(1): 1-12.
-
Hebbes CP, Thompson JP (2018) Pharmacokinetics of anaesthetic drugs at extremes of body weight. BJA Educ 18(12): 364-370.
-
Schetz M, De Jong A, Deane AM, Druml W, Hemelaar P, et al. (2019) Obesity in the critically ill: a narrative review. Intensive Care Med 45(6): 757-769.
-
Lewis TR, Shelton EL, Van Driest SL, Kannankeril PJ, Reese J, et al. (2018) Genetics of the patent ductus arteriosus (PDA) and pharmacogenetics of PDA treatment. Semin Fetal Neonatal Med 23(4): 232-238.
-
Flint RB, Ter Heine R, Spaans E, Burger DM, de Klerk JCA, et al. (2018) Simulation-based suggestions to improve ibuprofen dosing for patent ductus arteriosus in preterm newborns. Eur J Clin Pharmacol 74(12): 1585-1591.
-
Samura M, Takada K, Yamamoto R, Ito H, Nagumo F, et al. (2021) Population pharmacokinetic analysis and dosing optimization based on unbound daptomycin concentration and cystatin C in nonobese elderly patients with hypoalbuminemia and chronic kidney disease. Pharm Res 38(6): 1041-1055.
-
Song JC, Gao H, Qiu HB, Chen QB, Cai MH, et al. (2018) The pharmacokinetics of dexmedetomidine in patients with obstructive jaundice: A clinical trial. PLoS One 13(11): e0207427.
-
Ma X, Shang X, Qin X, Lu J, Liu M, et al. (2020) Characterization of organic anion transporting polypeptide 1b2 knockout rats generated by CRISPR/Cas9: a novel model for drug transport and hyperbilirubinemia disease. Acta Pharm Sin B 10(5): 850-860.
-
Paul A (2019) Drug distribution. In: Raj GM, (Eds.), Introduction to basics of pharmacology and toxicology. Springer, Singapore, pp: 89-98.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review