Therapeutic Monitoring-Guided Dosing of Factor Viii in Hemophilia A: From Pharmacodynamics and Pharmacokinetics toward Precision Therapy
Hemophilia is a rare hypocoagulation disorder that, depending on the lacking coagulation cascade factor, has different denominations. This review focuses on Hemophilia A (HA), particularly on the FVIII concentrates that are available in clinical practice to replace the scarce levels of FVIII observed in these patients. In fact, pharmacological responses are strongly heterogenic namely due to disease evolution and FVIII concentrates pharmacokinetic profiles. Therefore, therapeutic drug monitoring (TDM) is essential to maximize FVIII effectiveness and decrease adverse event rates. We provide a critical overview of current FVIII concentrates their mechanistic and pharmacokinetic differences as well as the factors that determine those profiles. Precision dosing through therapeutic drug monitoring is expanding and is essential in populations with altered pharmacokinetics and/or pharmacodynamics. However, there is still a need for studies correlating pharmacokinetics and patient outcomes. Herein, a pharmacokinetic-based optimization of FVIII therapy was revised and in deeply explained hot it can be successfully applied in clinical practice.
Hemophilia
The dynamic physiological mechanism named hemostasis is important to maintain the normal blood flow after blood vessels trauma [1]. It involves two coordinated systems: the procoagulant, encompassing the primary and secondary hemostasis to cease the blood loss through the thrombus formation and, the anticoagulant system, which includes the negative regulators towards the first system. Hemostasis disorders arise from the unbalance of these coordinated systems, leading to hypercoagulation diseases, caused by an anticoagulant deficiency, or hypocoagulation diseases, where the procoagulant activity is in deficit [1]. Hemophilia is acknowledged as the “royal family disease” due to the historic identification of Queen of England Victoria (1837 to 1901) as a carrier of the hemophilia gene and the death of her son, with 31 years old, with hemophilia and a brain hemorrhage [2].
Clinically, hemophilia leads patients to a continuous and uncontrollable bleeding [3] due to the underlying poor clotting activity. The faulty coagulation process is related to the deficiency regarding clotting plasma factors [4]. Depending on the missing clotting plasma factor, hemophilia is classified as Hemophilia A (HA), associated to the lack of Factor VIII (FVIII); Hemophilia B, related to the reduction of Factor IX (FIX) or, Hemophilia C, characterized by the defect on Factor XI (FXI) [4, 5]. Herein, HA will be focused on.
Hemophilia A
Epidemiology, incidence and prevalence: HA is a congenital, recessive X-linked disorder caused by the lack or deficiency of clotting factor VIII (FVIII). The severity of the disease depends on the reduction of levels of FVIII, which are determined by the type of the causative mutation in the genes encoding the FVIII, located in the long X chromosome arm, at Xq28 [6]. This gene is only expressed in male germline, in sperm cells, justifying the fact that men are the most affected while women are mainly carriers of the gene [3]. Generally, pathogenic mutations in FVIII gene lead to a dysfunctional FVIII protein as they can: (I) interfere with protein secretion, targeting the folding and intracellular processing; (II) reduce protein activation; (III) change the structure and stability of the cofactor FVIIIa; and/or (IV) promote abnormal interactions with serine protease Factor IX (FIXa) as then affect the tease complex [7, 8].
The most common form hemophilia A englobes 80 to 85% of the cases [8]. Data from the 2019 annual report revealed that the worldwide incidence of HA was 24.6 cases per 100,000 male birth patients while the prevalence, surrounded the 17.1 cases per 100,000 male patients [9]. Since that the current worldwide number of males is estimated to be 3.8 billion, the number of patients with HA is expected to be 794,000 [9].
Degrees of severity: The degree of severity of HA depends on the activity of the coagulation activity of the patient which, in HA, particularly regards FVIII. In HA, patient coagulation activity is represented by FVIII:C, where “C” is the plasma concentration at a corresponding time [10], and the values are expressed in international units (IU) or percentage (%), per dL or mL, which means that 1 IU is the plasma activity present in 1 dL (or mL) of plasma [11].
Healthy patients have a FVIII: C between 40 to 150 IU/dL whereas, HA patients exhibit values below this range [3, 12]. Depending on the residual FVIII activity, HA severity may be classified into, mild, moderate and severe (Table 1).
| Severity | Bleeding Manifestations | |
|---|---|---|
| 40-150 | Healthy | |
| May-40 | Mild | Bleeding with major trauma/surgeries |
| 01-May | Moderate | Bleeding with minor trauma/surgeries Occasional cases of spontaneous bleeding |
| <1 | Severe | Bleeding with minor trauma/surgeries Recurrent spontaneous bleeding in joints or muscles |
Table 1: Factor VIII levels associated to severity and bleeding occurrences.
Severe HA is described by recurrent and spontaneous bleeds with 90% of them starting at the joints (knees, elbows, ankles, shoulders and wrists) [12], 10 to 25% at the muscles (i.e. lower legs or forearms) [13], and 5-10% from other body organs [3]. Newborns and children with severe HA phenotype commonly experience soft tissues, intramuscular and mucocutaneous bleeding as well as extracranial and intracranial hemorrhage [3, 13]. The annual report from 2019 also offered predictions regarding the incidence and prevalence for severe phenotypes. Namely, the incidence was estimated for 9.5 cases per 100,000 males, whilst prevalence was 6.0 cases per 100,000 males. Globally, on those 794,000 patients with HA, 270,000 are severe [9].
Mild hemophilia patients are often under-diagnosed as they only bleed when triggered by a trauma or major surgeries. Furthermore, they are more sensible to bleeding in minor surgeries but they may even start to experience spontaneous bleedings [3].
Biological Mechanism of Factor VIII
FVIII is a glycoprotein synthesized by the liver sinusoidal cells, Kupffer cells and hepatocytes [14]. Structurally, FVIII is a complex protein [15] with an arrangement of: (NH2) A1-a1- A2-a2-B-A3-C1-C2 (COOH) [16]. In Golgi complex, it suffers proteolysis with two intracellular cleavages within the B-domain, resulting in a heavy chain with variable size (A1- A2-B domain) and light chain with constant size (A3-C1-C2 domain [16, 17]. FVIII is then secreted to the bloodstream as an inactive heterodimer, forming a noncovalent linking with the multimeric protein [17].
Coagulation cascade is activated upon a vascular damage (initiation phase) Figure 1, leading to the release of tissue factor (TF) in blood vessels, which, further, will bind to the activated factor VII (FVIIa) (Figure 1) [18]. The interaction between both factors is crucial to start the activation of the factor X (FX) and factor IX (FIX). The initiation phase ends with thrombin (FIIa) synthesized through the bound of activated FX (FXa) and activated factor V (FVa) (i.e. the prothrombinase complex) (Figure 1). The amount of FIIa is very small at this stage (approximately 2% of the required concentration) and, therefore, the coagulation process is ineffective to arrange a proper platelet plug [19].
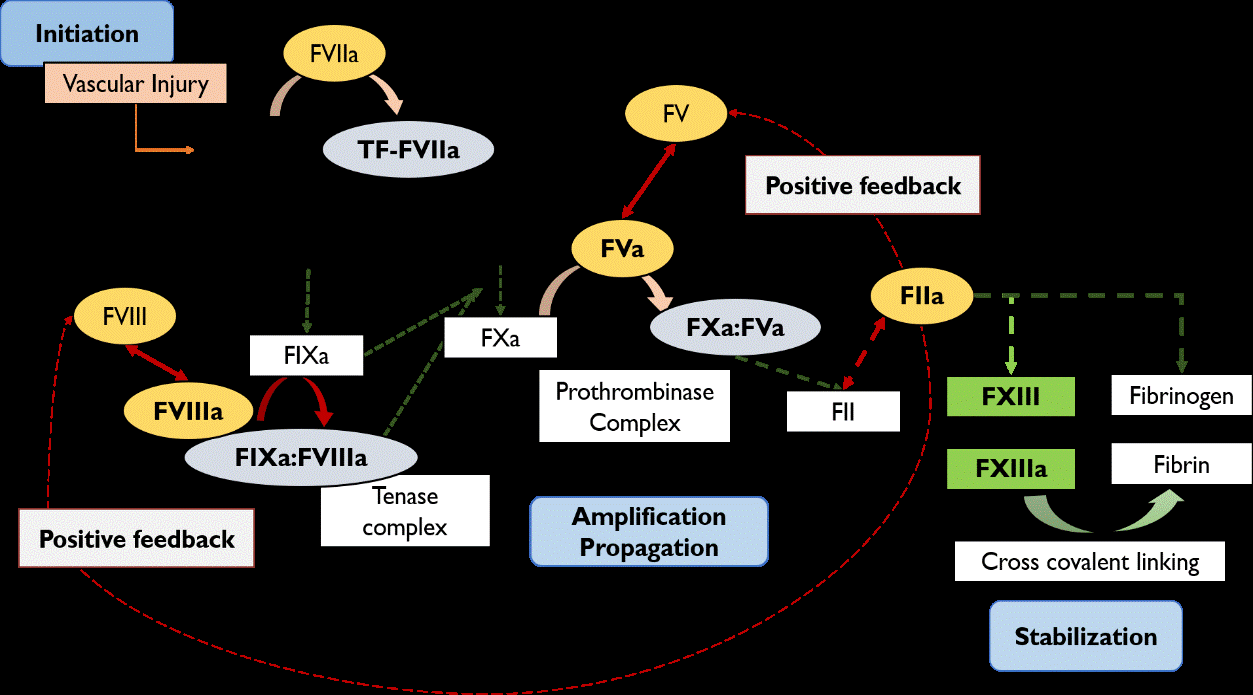
Figure 1: Mechanism of coagulation cascade. FVIIa, factor VII activated; FIX, factor IX; FIXa, factor IX activated; FX, factor X; FXa, factor X activated; FVIII, factor VIII; FVIIIa, factor VIII activated; FV, factor V; FVa, factor V activated; FII, prothrombin; FIIa, Thrombin; FXIII, fibrin-stabilizing factor; FXIIIa, fibrin-stabilizing factor activated; TF, tissue thromboplastin or tissue factor.
As a result, FIIa activates FVIII by interaction at heavy and light chains to dissociate FVIII from vWF [19, 20]. FVIII will then change its structure to a heterotrimer [17]. In parallel to FVIII activation, FIIa also targets factor V (FV), activating its cofactor (FVa) [19]. Consequently, FVIIIa interacts with FIX and form the tense complex (FVIIIa: FIXa), enhancing the activation of the FX whereas, FVa binds to the FXa (prothrombinase complex), producing more prothrombin (FII) and completing the amplification phase with this positive feedback response [19]. This mechanism guarantees a continuous production of FIIa at the surface of activated platelets (propagation phase) [18]. Furthermore, FIIa acts on fibrinogen to form the fibrin monomers [21], which are weakly connected. Therefore, FIIa activates the factor XIII (FXIIIa) to covalently link the monomers and held together a strong clot to cease the blood (stabilization phase) [19].
In brief, FVIIIa plays a critical role in the middle phase of coagulation (amplification/propagation) being an essential cofactor for the intrinsic tense complex and prothrombinase complex in order to increase the levels of thrombin and cease the blood loss with thrombus formation [17].
Clinical Relevance of Uncontrolled Factor VIII Levels
Factor VIII Protein Levels
Laboratory testing is essential to assess the clot rate formation upon activation of the coagulation cascade [22]. Accurate diagnosis for HA should be suspected when a patient, regardless of the age, presents a clinical medical history of easy bruising, spontaneous bleeding with no specific underlying reasons or, an excessive bleeding after any trauma or surgery [3]. In this context, the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) must be analysed (Figure 2) [22].
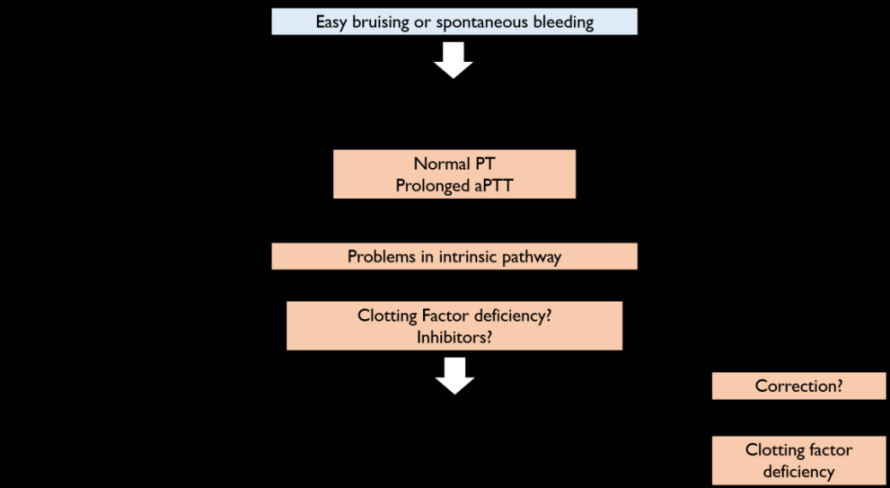
In inherited deficiency disorders, PT values are normal whereas aPTT are enlarged [22]. Additionally, a prolonged aPTT may be related to different clotting factors defects or to an immunologic response explained by the production of inhibitors as it will be discussed in Section 7 [23]. Therefore, mixing studies are the next step to accurately confirm the abnormal aPTT prolongation [22, 23]. These studies usually involve an equal volume (50:50) of the citrated patient plasma mixed with normal pooled plasma. If the aPTT becomes corrected means that a factor is indeed missing in the patient (i.e. hemophilia) [22]; if the aPTT is not properly corrected, other causes may justify the aPTT prolongation, namely immunologic patient response [23].
In a scenario where the deficiency is a result of a missing clotting factor, confirming tests are required to identify the lacking clotting protein. To attain this objective, the concentrations of each coagulation factor are tested and determined by one-stage or chromogenic assays, which are hence very useful not only to attain a definitive diagnosis of HA but also for drug therapy adjustment [11, 23]. One- stage assays are often used in clinical practice [3] where the patients’ citrated plasma is mixed with plasma FVIII deficient (levels <1 IU/dL) and compared to a standard reference, with known FVIII levels [22]. Results are represented with the clotting time in the y-axis and, the FVIII levels on the x-axis [24]. For instance, a FVIII: C value of 7% comparing to the standard plasma and, if this standard reference has a FVIII:C about 85 IU/dL, patient presents a concentration FVIII 6 IU/ dL (7% x 85) [25].
On the other hand, chromogenic tests use the patient plasma in mix with other coagulation cascade factors such as thrombin or prothrombin, factors IX and X, calcium and phospholipids in order to encourage the activation of FVIII and, subsequently will interact with factor X (FX) as reported in section 2 [26]. Here, the assay will measure the rate of FVIII to form the FX cofactor (FXa) by adding a chromogenic substrate (p-nitroanaline) that will reproduce a yellow color (due to specific affinity towards FXa) [25]. The emitted color is proportional to the amount of FVIII present in patient plasma [26].
Importantly, both methods have different sensibility and accuracy, with FVIII:C levels 15-20% lower with one-stage assays than with chromogenic tests. This has implications for interpretation of pharmacokinetic parameters, eventually with overestimation of the half-life [27]. Chromogenic assays are more accurate to detect FVIII: C between 0.1-2 IU/ dL [3] being hence, ascribed as the preferable method for monitoring clotting factor concentrates (CFCs) [27]. However, these assays are not always available in clinical practice and, hence, guidelines demand that pharmacokinetic studies are developed using always the same methodology in order to reduce data variability [3, 27].
Low Levels of FVIII
Extreme low FVIII levels (<1IU/dL) represent 60 to 70% of the HA patients [28], who, if not properly managed, experience approximately 15 to 35 spontaneous bleeding in the joints and muscles, per year [29]. Prolonged spontaneous bleeding in the joints, also referred as hemarthrosis, promotes the release of iron from hemoglobin, stimulating the production of cytokines and pro-angiogenic factors [28, 30], leading to an acute intra-articular inflammation named synovitis, and hypertrophy of synovium, called hemophilic synovitis [30]. The continuous environment of inflammation and hypertrophy induces a chronic and vicious cycle on the target joint that will progressively lead to bone damage, osteoporosis, and degeneration of the articular cartilage and atrophy of the muscles. Hemophilic arthropathy is the denomination for this final stage of the worst clinical outcome of the disease as patients experience extreme disability in their lives [30].
Another consequence is muscle bleeding [19, 28], specifically in pelvis muscle as it is difficult to control the loss of blood, and prolonged hematomas that lead to atrophy of the tendons, ossifications or hemophilic pseudotumor. Importantly, even though central nervous bleeding is less frequent to happen in hemophilia (<5%), it cannot be disregarded, due to its life-threatening. The simplest headaches for a long period of time or, a simplest somnolence, in HA may be an early diagnosis for intracranial bleeding [3]. If not properly detected, there is a strong probability for permanent neurological damage [29, 31].
High Levels of FVIII
In contrast to the low levels of FVIII, elevated levels are related to the increased risk for thrombosis, particularly the venous thrombosis [28, 32]. Several studies concluded that high FVIII levels (≥150 IU/dL) were observed in 57% of the patients with recurrent venous thrombosis, probably due to the increase of thrombin and fibrin rate or by inducing resistance to activated protein C, which is essential to down- regulate coagulation cascade [28, 32, 33].
On the other hand, arterial thrombosis is also observed with higher levels of FVIII. The explanation may rely on von Willebrand factor (vWF) which is increased due to forces in stenosis vessels, stimulating platelet adhesion/aggregation at the damage arterial wall or, the higher FVIII levels itself will increase thrombin formation and platelets activation [34, 35, 36].
In conclusion, uncontrolled levels of FVIII are associated to sub therapeutic or adverse effects that compromise efficacy and safety of the treatment and consequently life quality of the patient. Indeed, uncontrolled levels will increase patient outcomes such as chronic pain, disability, society deprivation associated with anxiety and depression, all subscribed for a negative impact in quality of life. Therefore, a proper control of FVIII levels is crucial in clinical daily practice [37].
Treatment Management
Over the years, HA treatment suffered substantial improvements. The first therapeutic option was available in 1840 with blood transfusions [38]. From 1950 to 1960, patients were treated with fresh frozen plasma but the amount of FVIII was not enough and, hence, patients either died at early ages or, lived longer with reduced quality of life [39]. The year of 1964 was marketed by the discovery of Judith Pool, who attained higher levels of FVIII in thawing plasma, allowing better care in HA [2]. Years later, plasma- derived FVIII (pdFVIII) concentrates were the first home treatment, offering a better control in bleeding as well as an improvement in patient’s quality of life. Later, mild severity gained an effective, safer and cheaper treatment, the desmopressin (DDAVP) [40].
However, treatment journey was not always a golden path as, in 1980, a “dark era” was witnessed, after several patients prescribed with pdFVIII, were infected with human immunodeficiency virus (HIV) or hepatitis C virus (HCV). Most of these patients either died or lived with severe sequels [40]. In parallel with the development of new viral inactivation steps, DNA technology was exponentially increasing contributing for the important cloning of the FVIII gene [2]. As a result, it was possible to reproduce the FVIII protein in mammalian cells by recombinant DNA technology. These new drugs, named recombinant concentrates (rFVIII), had their efficacy proven only in 1989 and the first launch dated 1992 [39]. Since then, rFVIII also had their improvements allowing for their categorization into four generations.
Furthermore, innovations did not stop in rFVIII area and, since 2010, there has been an interest in gene therapy as it is a more specific and accurate to stimulate the body to synthetize the missing protein. As an opportunity to improve more the quality of life of HA patients, this still being a clinical unmet need.
Therapeutic Regimens
In HA, the available regimens are distinguished by the final purpose of the treatment. Thus, in episodic bleeding, patients are treated under the on-demand regimen whereas, prophylaxis regimen is important to prevent the worsening of clinical outcomes [3]. The on-demand approach consists on the administration of the CFCs only when the bleeding episode starts; its objective is reduce pain and manage the acute impact of the bleeding [3, 41]. Since its application is recommended only at the start of an hemorrhagic episode, the chances of a small bleeding turning into a larger hemorrhage and, subsequently, worst clinical outcomes are high [41].
Prophylaxis regimen emerged when it was discovered that less hemorrhages and less cases of hemophilic arthropathy occurred in moderate HA patients [42]. Therefore, it was mandatory to stablish prophylaxis as a regimen that maintained FVIII levels above 1 IU/dL to convert the severe stage into moderate/milder stage and, hence, experience less HA symptoms [43]. An effective prophylaxis involves regular intravenous infusions of CFCs in order to prevent and preserve the musculoskeletal function and to allow a normal life-style and a better quality of life [3].
Prophylaxis is subdivided in three classes (primary, secondary, tertiary) based on the moment that the treatment management was initiated Table 2. When the patients start the prophylaxis early in life (i.e. primary and secondary) the chances of getting better long-term outcomes are higher. Hence, in clinical practice, this is seen as a goal standard choice [3, 44]. In contrast, later prophylaxis will only reduce the pain and inflammation as well as slowing down the progression of HA [3, 44]. The intensity of prophylaxis depends on dose and frequency of administration (Table 3) [3]. The Swedish prophylaxis (or the Malmö protocol) is the most intense regimen and it is characterized by the administration of 25 to 40 IU/kg of FVIII every two days for, at least, three times per week [45]. The Dutch regimen (or Utrecht protocol) is of intermedium intensity once patients are prescribed with 15 to 30 IU/kg, two or three times per week to avoid spontaneous bleeding [44, 45]. The lowest intense regimen is considered when the dose varies between 10 to 15 IU/kg for two or three days per week [3].
| Class of prophylaxis | Age of initiation | Expected clinical history |
|---|---|---|
| Primary | <3 years old | Physical exams and/or imaging test with no joint disease Before the second evident joint bleed |
| Before the onset of a joint disease After two or more joint bleed | ||
| Secondary | ≥3 years old | |
| After a documented joint disease | ||
| Tertiary | Any age; mostly adults |
Table 2: Type of prophylaxis by the age of initiation.
| for header rows and for data cells. Use for bold text within cells. Preserve empty cells. Return ONLY the HTML , no other text. | |||
|---|---|---|---|
| Regimen | Intensity | Advantages | Disadvantages |
| Swedish (Malmö protocol) | High | Guarantees minimum levels of 1 IU/dLLower annual joint bleedsBetter long-term joint outcomesGreat for active lifestyle patients | Adherence (more infusions)Expensive (more doses)High over treated mild phenotypes |
| Dutch (Utrecht protocol) | Moderate | Less expensiveMore quality of life than low intensityReduce chances of bleeding to 90%Low annual joint bleeds (1 per year)Good for adolescents and adults | Undertreated patientsSlightly worse long-term musculoskeletal outcomes |
| Low intensity | Less expensive (many countries can afford)Reduce bleeding incidence versus on-demand by 80%Reduced annual joint bleeds to 3 per year | Unknown long-term effect on musculoskeletal outcomes |
Table 3: The advantages and disadvantages of each prophylaxis intensity regimen in HA.
In alternative to these three main protocols, it is possible to start with the low intensity of once weekly infusion and, then, escalate administration frequency [3]. Named Canadian protocol, this regimen is particularly important to enhance treatment compliance in young children [3].
Factor VIII Concentrates
Since HA patients do not synthesize enough FVIII, the rational of the treatment is to replace the FVIII levels by administration of CFCs, which includes plasma-derived (pdFVIII) and recombinant products (rFVIII) (Figure 3). However, these drugs are expensive and may not be supported in poor health systems, which instead may use cryoprecipitates or fresh frozen plasma (Figure 3), even though these are not submitted to a safety inactivation procedure like pdFVIII. Therefore, patients have a high probability of developing infections [3].
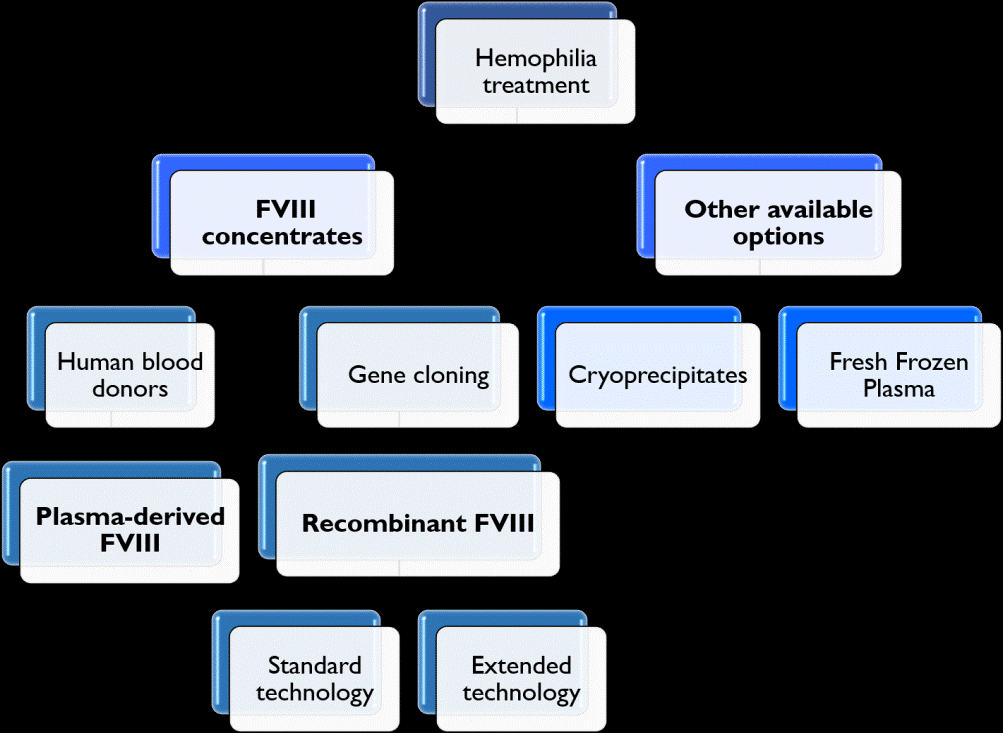
Plasma-Derived Concentrates
Plasma-derived FVIII concentrates are available since 1970 [40] and they are obtained by cryoprecipitation, which is the result of a precipitation process at cold temperatures [46]. As these concentrates are derived from plasma donors, the aforementioned contaminations only happened due to the lack of viral purification steps within the manufacturing process [40]. Since then, the scientific community and pharmaceutical industries improved the safeness of pdFVIII by introducing viral inactivation techniques such as dry heat, pasteurization, vapor heat and solvent/detergent (Table 4) [47].
| Trade Product Name | Year of approval | Manufacturer | Product Characteristics | ||
|---|---|---|---|---|---|
| Active Substance | Viral Inactivation | Viral purification | |||
| Emoclot | 1999 | Kedrion S.p.A. | FVIII | S/D Dry heat | Ion exchange chromatography |
| Fanhdi | 2001 | Grifols | FVIII | S/D Dry heat | Heparin ligand chromatography |
| Octanate | 2015 | Octapharma | FVIII | S/D | Ion exchange chromatography |
| HaemateP | 2000 | CSL Behring | FVIII | Pasteurization | Multiple precipitation |
| +vWF | |||||
| Wilate | 2012 | Octapharma | FVIII | S/D | Ion exchange chromatography |
| +vWF |
Table 4: Summary of plasma-derived concentrates characteristics available. FVIII: factor VIII; S/D: solvent/detergent; vWF: von W
In the past, dry heat treatments consisted on submitting the pdFVIII concentrates to temperatures between 60-80ºC, for 24 to 96 hours, only inactivating the HIV [46]. Since then, alternatives have emerged, and products are now either submitted to higher temperatures (80ºC – 100ºC, for 72 hours or 30 minutes respectively), targeting a wide variety of virus, including HIV, HCV and hepatitis A virus. On the other hand, pasteurization has revealed to be a highly effective method as the drug undergoes for heat treatment (60ºC), for 10 hours, in the presence of the FVIII stabilizers such as sugars, amino acids, or acetate, to prevent loss of activity [46, 47]. Similarly, vapor heat consists on applying water vapor before heating the product to 60ºC for 10 hours [47]. Finally, the solvent/detergent technology is the most effective for lipid membranes of certain virus [46]. It consists in a mix of organic solvents (e.g. tri-n-butyl-phosphate) and detergents (e.g. Tween-80, Triton X-100) that target virus membrane and, consequently, inactivate them. It is effective against HIV, HBV, HCV, West Nile virus, Dengue virus and Zika virus.
Alongside with viral inactivation procedures, pdFVIII concentrates go through purification by chromatographic methods that separate the viruses from the protein [48]. There are several variants of chromatography but the most used on pdFVIII purification are the affinity chromatography and the ion exchange chromatography [46]. The difference between them regards the molecules that are used to separate the components. Affinity chromatography is more specific and use ligands such as heparin, metals or gelatin while the ion exchange uses electric charged molecules [46]. Prior to formulation, pdFVIII are filtered to remove smaller viruses that can be present in the product [47] and the most frequently usedmethod is nanofiltration with filters pores ranging from 35 to 15 nm, retaining the virus on the nanofilter [46, 47].
A side but important note regards the possibility of the contamination and subsequent transmission of prions within pdFVIII. Prions are associated with fatal neurodegenerative disorders like the Creutzfeldt-Jakob disease [47]. They are resistant to inactivation procedures [46] and therefore the only steps that have been proved to be efficient are precipitation, chromatography and filtration [47].
Recombinant Concentrates
Cloning was a big step for HA treatment. Recombinant FVIII concentrates consists on the heterologous transfection of the FVIII DNA plasmids into a cell line that is, then, cultured and stabilized by plasma proteins derived from humans or animals [49]. In contrast with the previous products, rFVIII are more safer, justified by the significant reduction of the transmission of viruses and/or prions [49, 50].
The concentrates evolved over the years in terms of the manufacturing methods and, on the incorporated technology to gain more efficacies in bleeding control. Four generations were created to distinguish each drug. Furthermore, these generations are only validated to the first manufactured products, the standard half-life (SHL) while, the extended half-life (EHL) ones are the recently innovations formulated to prolong the rFVIII activity [49].
Standard Half-life
First generation: The first SHL product was launched in 1992 by FDA, named as Recombinate®, also known as Antihemophilic Factor [7]. This product was cultured in the non-human cell line [Chinese Hamster Ovary (CHO) cells], using animal proteins in medium culture (e.g. bovine-insulin, -aprotinin and -albumin) and, human albumin as stabilizer [51]. For viral safety, affinity chromatography by a monoclonal antibody (immunoaffinity) was introduced alongside with ion exchange chromatography; however this first generation had a reported risk of transmission of nonenveloped viruses and prions associated to the Creutzfeldt–Jakob disease [39] and, hence, the pharmaceutical industries started to improve their manufacturing process by developing new generations of rFVIII.
Second generation: The second generation of SHL products is characterized by the use of human proteins in medium culture (e.g. human serum albumin) instead of the animal- derived ones and, by replacing albumin for sucrose as formulation stabilizer [39]. Kogenate Bayer® (also marked as Kogenate FS® outside of Europe Union) is a second generation drug, using octocog alfa as the active substance and Baby Hamster Kidney (BHK) as the cell line used to express the FVIII [51]. Moreover, in viral purification, S/D and filtration were coupled to chromatography to guarantee the inactivation of the viruses [7].
Helixate NexGen® (marketed as Helixate FS® outside of Europe Union) was also part of this generation, but it was recently withdrawn from the European Union as requested by marketing authorization holder, Bayer AG.
Third generation: The third generation emerged aiming to recreate rFVIII drugs with reduced chances of virus transmission through the loss of the animal or human- derived proteins throughout the manufacturing process [51]. This is the most extensive generation so far introduced in the market and the most currently prescribed in clinical practice.
The first drug fitting into this category was ADVATE® using the same active substance as the previous generation (octocog alfa) but the CHO cell line [51]. The drug proved efficacy in controlling and preventing bleeds as prophylaxis, with 88.5% of successful rate, by infusing only one or two doses [52]. The annual bleed rate (ABR) for standard prophylaxis (25–40 IU/kg, 3– 4 times a week) was 6.0 whereas, on-demand, it was 18.5. This supports that ADVATE® is more efficient in patients subjected to prophylaxis regimen [28]. Importantly, ADVATE® has demonstrated to be safe with no inhibitors detected [52].
Refacto AF® (moroctocog alfa) emerged as the first product with B-domain deleted (BDD) from the structure of FVIII [51]. Removing this domain of the mature protein demonstrated that, upon its delivery into the bloodstream, the FVIII molecule is activated by thrombin (FIIa) and the conformational structure changes for an heterotrimer with no B-domain involved [17]. Therefore, the removal of BDD does not interfere with FVIII function. Indeed, it enhances the secretion even more, as it has been demonstrated by the higher levels (i.e.17-fold higher) found for FVIII mRNA [53]. Regarding its efficacy, moroctocog alfa prevents spontaneous bleeding in a defined prophylaxis routine, especially in patients with history of target joints [54]. Furthermore, it is effective in prophylaxis regimen (30 IU/ kg; three times per week) in 94 patients as 60.6% of them did not experience spontaneous bleedings, contributing for a low annual bleeding rate (ABR=3.9). Safety of moroctocog alfa has also been proved in clinical trials with no significant immunogenicity response [54].
NovoEight® (turoctocog alfa) is another third generation rFVIII with improvements at the B-domain [55]. This drug uses the B-domain truncated (BDT) and, even though the B-domain is not essential for its activity, it is usually highly glycosylated as a result of post-translational changes (N-linked glycosylation and O-linked glycosylation). This allows a proper intracellular transport and subsequent processing of the FVIII protein [53]. Moreover, the full- length structure drugs have, usually, nineteen N-linked glycosylation, making them more difficult to express the FVIII [56]. Therefore, turoctocog alfa has the advantage of being expressed more easily due to the truncation of the respective domain as it has only four N-linked glycosylation (two in A1 domain, one in A3 domain and one at C1 domain) [55] and one O-linked glycosylation at the B- domain (Ser750) [49]. Its efficacy and safety were investigated in adults and adolescents, in the GUARDIANTM 1 trial enrolling 150 patients [57]. Accordingly, turoctocog alfa showed to be effective in bleeding control with one to two infusions. Patients had an ABR of 3.7 bleeds per patient per year under prophylaxis regimen [57]. In terms of safety, it was hypothesized that it would have a bigger immunologic response due to the engineered B-domain truncated (BDT) [55]. However, the clinical safety data showed no concerns of this matter as none of the patients enrolled in the study developed inhibitors [57].
Kovaltry® (octocog alfa) was based on Kogenate Bayer® but adding new features on its technology [58]. The remain characteristics are the amino acid sequence, the full-length structure and the cell line chosen to express FVIII (i.e. BHK). The innovations started with the addition of the human heat shock protein 70 (HSP70) gene, an intracellular chaperone that will ensure the proper folding of FVIII and, consequently, increase the protein expression [58]. Moreover, the N-terminal glycans present 96% of sialic acid (i.e. sialylation) which seems to be responsible for the 10% prolonged half- life (12.2h versus 13.4h) and slower clearance (0.043 versus 0.036 dl/h/kg) of Kovaltry® comparing to Kogenate Bayer® [7]. Clinical data was evaluated in the LEOPOLD clinical trial to assess the efficacy in 62 patients aged from 12 to 65 years old [58]. Among them, 44 patients (71%) were treated prophylactic three times per week and 18 patients (29%) treated prophylactic twice per week. No major differences were verified in ABR for each patient group as the median of total bleeds were low (1.0 for twice times/week regimen and 2.0 three times/week). As for safety, no immunologic response was observed in previous treated patients and no serious adverse events were observed [58].
The last third generation drug that was introduced in the market was Afstyla® (lonoctocog alfa), which is obtained applying the most unique technology. It is a single chain with the truncated B-domain that serves as a linkage between the heavy chain and the light chain [59]. The rational is that, endogenous FVIII have both chains connected by a noncovalent divalent metal ion (Ca2+ or Mn2+) [8], which is easily to dissociate and becoming inactive [60]. Therefore, lonoctocog alfa is covalently linked by a BDT enhancing the stability and increasing the chances to interact with vWF and, subsequently, prolong the half-life comparatively with the full-length molecules [59, 61]. Clinical data of lonoctocog alfa was assessed in the AFFINITY program [62] and evaluated the short-term safety after a single dose (50 IU/kg) of lonoctocog alfa, in previous treated patients with severe HA [49]. Efficacy was evaluated on the second and third part of the trial, and lonoctocog alfa showed to be as efficient as the aforementioned rFVIII drugs in bleeding control (93.8%) with either one or two doses (median dose of 31.7 IU/kg). Low ABR values (1.14) were reported and no adverse events or inhibitors were detected [49].
Fourth generation: This generation is only described by Nuwiq® (simoctocog alfa) [63]. The difference between the previous generations refers to the use of human embryonic kidney cell line (Hek293) to express the FVIII [39]. The previous cell lines, CHO and BHK, have been described as a potential source for an immunologic response due to the presence of glycans epitope N-glycolylneuraminic acid (Neu5Gc) and Galα1→3Gal groups that are not present in the human form of the protein hence, being a source for the activation of the immune system [7]. Furthermore, using the human cell line has the advantage to mimic the endogenous FVIII, particularly, in post- translational modifications such sulfation [63]. Sulfation occurs in Golgi apparatus and it is important for FVIII function [53]. This modification targets the tyrosine residues (Tyr) located near to the acidic domains of the structure and all of them (six in total) are crucial for the activity of FVIII [53].
Moreover, it has been demonstrated that the sulfation of Tyr 1680 is responsible for the binding of vWF to FVIII, conferring more stability and protection against early degradation/elimination of the bloodstream [64]. The other drugs, from second to third generations, have the Tyr 1680 sulfated as well but in less proportion (1% to 6.5% in second-generation and 15% in third-generation). On its turn, simoctocog alfa has every tyrosine fully sulfated including the Tyr 1680 [63], decreasing patient immunogenicity and improving drug residence time in the bloodstream.
Extended half-life
Over the years, the SHL products were the best option to manage HA patients. However, these drugs deliver the active substance for a short period of time (8h to 12h) [64], obligating to a more frequent infusion (3 to 4 times weekly) to maintain the minimum levels of FVIII activity. This can justify the poor compliance in some cases [65]. Therefore, since 2010, new drugs have been developed to increase their half-life time, and hence require a lower frequency of infusions. This is the field where the extended half-life (EHL) started.
Today, an EHL drug must conquer three mainstream features. The first implies the use of an innovative engineering technology to extend drug half-life time. The second makes use of the bioequivalence cut off limits (80%-125%) to compare drug systemic exposure (given by AUC) between SHL and EHL drugs. If the ratio between the novel product and the standard SHL is above those limits, it is expected that we are working with an EHL product. Finally, the half-life time ratio extension needs to be of at least 1.3 higher for the new compound [58].
Based on these criteria, lonoctocog alfa was excluded as, when compared with ADVATE_®_, the AUC ratio was below the 125% (not “biodifferent”) and its half-life was only extended 1.09 h [66]. Currently, EHL are obtained through innovative technologies, that may lay on chemical modifications (PEGylation) or fusion with Fc domains of serum proteins with long half-life times (Fc fusion) as it will be explained in the next sections. PEGylation: PEGylation is a chemical modification that consists on a covalent bound between polyethylene glycol (PEG) molecule to FVIII [66]. The advantage of this conjugation is that PEG serves as a “shield” from the clearance receptors (prolonging drug half-life) and the immunogenic epitopes that reduce the immunogenicity [67].
The first product was Adynovi_® (Adynovate®_ outside the EU) using rurioctocog alfa pegol as the active substance and CHO cell line to express its activity. The design involved the full-length molecule ADVATE®, shield with a weighted 20 kDa PEG molecule. It demonstrated to be efficient in controlling bleeding episodes with prophylaxis (40-50 IU/ kg) since the ABR value was lower (median 1.9) [64].
Similarly, Jivi_® (damoctocog alfa pegol) is expressed in BHK cells and uses the B-domain deleted structure linked to a single 60 kDa PEG molecule through an amino acid substitution by cystine [64]. Esperoct®_ (Turoctocog alfa pegol) is the last and the most recent EHL introduced in the market. It englobes the B-domain truncated molecule (turoctocog alfa) glycoconjugated to a PEG substance of 40 kDa [7], expressed in CHO cells [64]. Glycoconjugation means that, the PEG molecule is place, via enzymatic, in one of the o-linked glycans at B-domain [7]. Fc Fusion: The Fc domain of immunoglobulins establish fusions with other molecules of the body such as, cytokines or growth factors [68]. Endothelial cells express the neonatal Fc receptor (FcRn), at the same site where IgG coexist to protect the vasculature. The fusion between both components (i.e. FcRn and Fc domain of IgG) is documented at epithelial cells of certain organs (e.g. lungs, kidneys, intestine). In addition, studies proved that, FcRn protects IgG from lysosomal degradation on the vascular endothelium, recycling it back to the bloodstream [68, 69], prolonging, hence, the half-life time up to 21 days (Figure 4).
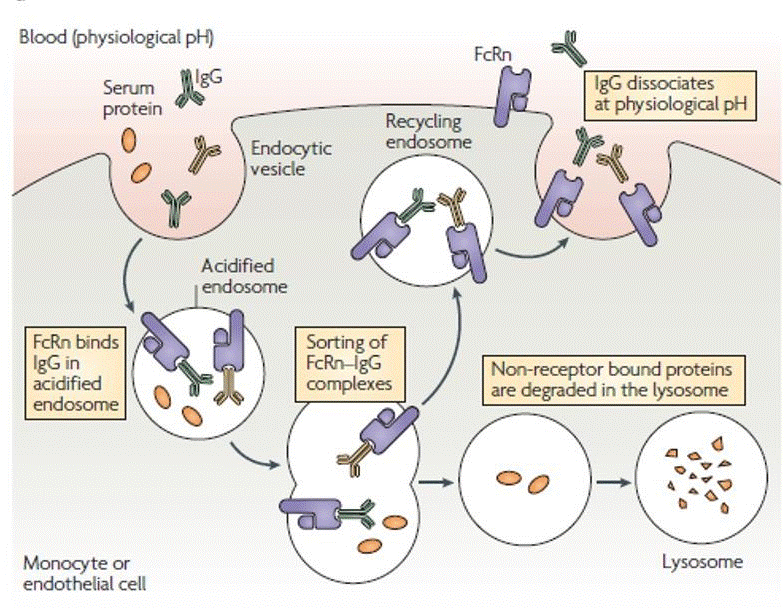
Therefore, this mechanism was replicated to HA treatment as a new opportunity to prolong the FVIII life on the bloodstream and enhancing his activity. Elocta_®_ (Efmoroctocog alfa) is the only product currently available which links covalently the Fc portion of IgG1 to the molecule. It has the BDD and is expressed in Hek293 cells [68].
Immunogenicity
Development of antibodies against CFCs was firstly reported in 1940 by Lawrence that described them as neutralizing alloantibodies [70]. They are an immunologic response to the treatment with CFCs [71], making the treatment ineffective and promoting a higher susceptibility for bleeding episodes. Neutralizing alloantibodies are currently designed as inhibitors of FVIII. They have a high affinity to certain epitopes present on A2, C1, and C2 FVIII domains [72], interfering with the FVIII either by blocking its mechanism (restrict the binding sites for FIX, phospholipids, and vWF) or removing it from circulation (i.e. enhanced clearance) [71, 72]. Moreover, antibodies are not only “neutralizing” towards FVIII. Indeed, patients can synthesize “non-neutralizing” or “non-inhibitory” antibodies. Although they do not express a function directly on FVIII activity [72], it has been reported their impact on the catabolism [73] and pharmacokinetics of CFCs [74]. These types of antibodies may be important as biomarkers for the neutralizing antibodies, after one study discovered positive inhibitors 1.5 years later in patients previously with non-neutralizing inhibitors [75].
Besides these two categories, the immune system can also develop auto-antibodies in non- hemophilia patients, as a condition named acquired hemophilia. It is a very rare condition with an incidence of about 1 case per million people per year [76]. Half of the patients diagnosed with acquired hemophilia usually are related to clinical conditions such as autoimmune disorders, tumors or to the postpartum as a rare adverse event (risk between 7% to 21%) [77]. In addition, age can also explain the idiopathic cases, particularly in elderly as they are a more vulnerable population [76].
Screening of the Inhibitors
Inhibitors detection is possible using either the Bethesda assay or the modified version, the Nijmegen Bethesda assay, which is more sensitive and specific [3]. Both assays measure the concentration (also named as titer) of inhibitors [71]. Results are expressed in Bethesda Units (BU) meaning that, 1 BU is the equivalent inhibitor amount in 1 mL of human plasma that neutralizes FVIII by 50% [72]. To consider a positive result, the titer must be higher than 6.0 BU [3].
Furthermore, inhibitors should be screened before they interfere with the efficacy of the treatment and, preferably, when there is a high risk for their development, which is within the first 20 EDs. In children, they should be screened within the range of 5–20 EDs, and then every 10 EDs until 50 EDs. Afterwards, screening inhibitors should be performed twice per year until 150 EDs since the risk is much lower. In adults, normally the risk is lower and should be considered: (a) after intensive treatments; (b) before undergoing a major surgery; (c) clinical response to the treatment is suboptimal [78].
As for non-inhibitors, they cannot be detected with the previous gold standard assays. Instead, enzyme-linked immunosorbent assay (ELISA) or fluorescence-linked immunoassay is recommended [3].
Characterization of the Inhibitors
HA inhibitors are classified in two categories: the ones that are genetic (unmodified ones) or not related to genetics (environmental/modified) [79]. Regarding their structure, inhibitors are polyclonal immunoglobulin G (IgG), often within IgG4 or IgG1 subclasses [76]. According to their of peak activity, inhibitors are classified as low-titer inhibitors (<5.0 BU) or higher-titer inhibitor (>5.0 BU), both requiring different managements [3]. Low-titer inhibitors (LTI) often belong to IgG1 subclass and tend to disappear spontaneously after 6 months without need of management, which is why they are often described as transient inhibitors. Nonetheless, patients with this type of inhibitors should be closely monitored, every 2-4 weeks, because LTI can easily convert into higher-titer inhibitor (HTI) [78].
In contrast, HTI usually belong to IgG4 subclass and are persistent inhibitors [78]. This means that after a long period without a CFCs exposure but after 3-5 days of CFCs re-introducing, their response may increase (i.e. anamnestic response) [3]. These inhibitors are undetectable, creating resistance to the CFCs [78].
In addition to their activity response, inhibitors express different kinetics. Type 1 inhibitors act as a second-order kinetics (i.e. dose-dependent inhibition), fully inactivating FVIII activity; while type II inhibitors have a more complex kinetics with only a partial inactivation of FVIII activity. The prevalence of type 1 is observed in severe HA patients whereas type II is more frequent in mild or acquired hemophilias [71, 76].
Prevalence and Incidence of Inhibitors
Incidence is related to the number of the new inhibitors in HA cases over a period of time [78, 80]. Usually, in severe HA the incidence rounds the 30% whereas in moderate/mild HA it is approximately 3-13% [80]. In severe HA, prevalence is 5-10% which means that, at any time, approximately 5-10% of the patients with severe type will present inhibitors. However, this is influenced by the incidence rate, the type of inhibitors found (HTI and LTI), eradication with immune tolerance induction programs and the deaths related to inhibitors [78]. For non-inhibitors, studies have been reported a prevalence of 2-3% in healthy individuals but, higher values for hemophilia patients and a wide range related to the severity of the disease (12% to 54%) [81].
Management of Inhibitors
Upon detection of inhibitors and their classification as LTI or HTI, it is crucial to trace a proper management plan. Generally, if the inhibitor is LTI, it is possible to use porcine recombinant FVIII (prFVIII), usually prescribed for patients with acquired HA [78]. In 2015, the European Medicines Agency authorized the Obizur_®_ (susoctocog alfa), a prFVIII, which is a high-purity B-domain deleted structure manufactured by recombinant technology in BHK cells [82]. Desmopressin (DDAVP) is a synthetic vasopressin analogue with proved efficacy towards mild HA patients [83] as well as an option for LTIs when displaying a type II kinetics [78].
As aforementioned, LTI can easily turn into HTI, which promotes the use of bypassing agents such as plasma- derived activated prothrombin complex concentrates (aPCC) and recombinant factor VII activated (rFVIIa). Both have an efficacy of about 80-90% for bleedings management in patients with inhibitors [84].
Feiba_®_ (Factor Eight Inhibitor Bypass Activity) is an aPCC with viral inactivation process containing zymogens, factor II (FII), factor VII (FVII), factor IX (FIX), factor X(FX) as well as their activated forms (FIIa, FVIIa, FIXa and FXa) which help to restore hemostasis. Its recommended dose is 50-100 IU/kg with 200 IU/kg the maximum dose per day [78]. NovoSeven® (eptacog alfa) is a rFVIIa [78]. Pharmacologically, activated factor VII (FVIIa) is not enzymatically capable of activating itself which means that FVIIa needs a partner, the tissue factor (TF), to form a stable complex [85]. This complex quickly activates factor FXa, generating enough quantity of thrombin, which is crucial to activate the cofactors FVIII and FV. Another advantage of rFVIIa in terms of its mechanism of action is that, FVIIa is not easily inactivated by antithrombin so it is possible to establish TF: FVIIa complex without neutralizers [85].
Bypass agents are effective but prophylaxis with them, in a long term, is not cost-effective as the efficacy decreases as the morbidity risk increases [86]. Hemlibra_®_ (Emicizumab) became a new hope as it is the first non-factor replacement therapy administered subcutaneously prescribed for inhibitors management [86]. Emicizumab is an humanized bispecific monoclonal antibody (IgG1) that binds to FIXa and FX mimicking the function of FVIII [86, 87]. Contrary to the previous drugs, it has a long half- life (approximately 27 days) and no structural similarities to FVIII which may be the reason to work in these patients since it does not induce or enhance inhibitors development [86].
Switching
Switching is a clinical decision made by health care professional in which is suggested a swap from one concentrate to another [88]. This exchange may be between different category of concentrates (i.e. pdFVIII to rFVIII) or, within the same type but different technologies (SHL to EHL), which is more common [89]. This is a multifactorial decision (Table 5) that reunites several clinical concerns and expectations emerged from the patient or from the health professionals. Furthermore, switch is an individual assessment where the benefits and potential risks must be balanced [90].
| Major Concern | Clinical Expectations | |
|---|---|---|
| Patients | Safety | Avoid side effects: Hypersensitivity/allergy |
| Patients | Quality of life | Possibility to increase physical activity. Participation in society Good control and protection with bleedings Less infusions /Less venipunctures |
| Patients | Economics | Cost-savings |
| Health Care Professionals | Type of concentrate | Safety Efficacy Patient future compliance Assessment which new regimen is the best |
| Health Care Professionals | Pharmacokinetics | Initial parameters assessment Monitoring behavior Tailoring new regimen |
| Health Care Professionals | Immunogenicity | Medical history Monitoring long-term |
Table 5: Summary of the possible clinical reasons behind the switch [88,90].
Regardless of the type of concentrate chosen, the switch be considered when the efficacy is impaired (i.e. patients experience frequent bleeds and/or have target joints) [90]. Under these circumstances, it is more frequent to swap from a SHL concentrate to EHL since; this latter technology presents advantages regarding drug compliance and quality of life. Moreover, EHL is recommended in patients that are not able to prophylaxis with SHL or, for patients who are non-adherent for the treatment [90]. On the other hand, patients should not be submitted to a switch if, they are not experiencing breakthrough bleeds (or they are minimal) or if the management of the regimen (on-demand or prophylaxis) has no issues related [90].
Guidelines provide some recommendations for this clinical decision, advising professionals that only patients with more than 150 exposure days (EDs) with no prior inhibitor drug history should be considered [91]. The choice behind the number of EDs is explained by the vulnerability of developing inhibitors in patients with less than 50 EDs, enhancing their higher risk [90].
After patient evaluation and the reasons for a switch, the ideal regimen and the best drug must be decided. Trial- and-error is a common approach where two scenarios can happen: either the dose is kept the same and only the frequency is adjusted, or the dose and frequency from pivotal data studies are used [89]. However, it is important to bear in mind that CFCs have high inter-patient variability which means that, each patient will respond differently so, this approach is a risk to underdoing and subsequently, for more bleeding [89, 90].
Moreover, after drug switch, pharmacokinetics must be evaluated and tailored for a better clinical response [89]. It has been described that pharmacokinetics assessment can be performed after single-dose infusion or, in steady-state conditions which are attained after the administration of several doses [90]. Generally, trace a personalized regimen involves a definition of trough optimal levels to overcome the bleeds and target joints [90]. If the aim is to maintain the previous dosing regimen, switch aims at attaining high trough levels, whereas, if the dosing interval is enlarged to improve adherence, then trough levels must be maintained, and time spent at a lower factor level is considered [90].
The pharmacokinetics parameters must be adjusted to each situation as explained in deeply in section 8. After achieving the right regimen for each patient, they must be monitored for 10 EDs, 4 weeks and 3 months. Clinical evaluation focus on microbleeds, joint progression (bone density and structure), diagnosis of possible inhibitors formation and testing in a long-term neurological impairment if patients were switch to a PEGylated drug [92].
Pharmacokinetics of FVIII concentrates
Absorption, Distribution and Elimination
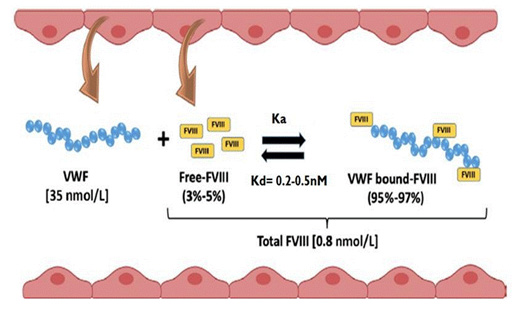
CFCs are administered by intravenous route and therefore their bioavailability is 100% with no occurrence of drug absorption [93]. The distribution process is influenced by the content of human body fluids and plasma proteins (e.g. albumin) [94]. Regarding FVIII CFCs, their action is exerted on the bloodstream [95]. Nevertheless, most of them distribute into the extracellular space (specifically at the intravascular compartment) due to their large molecular weight [10, 93]. Moreover, FVIII has high affinity to bind noncovalently and reversibly to vWF, creating a well balance complex that properly regulates the amount of the free form (i.e. in circulation) and, the one bound to vWF (i.e. as a complex). The literature describes FVIII plasma therapeutic concentration around the 0.8 nmol/L whereas, for vWF, it is approximately 35 nmol/L [96]. As vWF concentration is higher, it is expected an excess of 50 molars in bloodstream, at steady-state conditions [96], representing a FVIII molar ratio per vWF monomer of 1:50 [14]. In addition, the dissociation constant (Kd) is estimated to be between 0.2 and 0.5 nm which means that the affinity between both molecules is very high (Figure 5) [97] and most of the FVIII is linked to vWF (approximately 95 to 97%) [96, 98]. Thus, since the vWF serves as a shield for FVIII elimination, a faster elimination of FVIII is expected when it is as the free form (estimated half-life time: 2 hours); when complexed, FVIII elimination is slower (estimated half-life time: 12 hours) [96].
Elimination of CFCs is quite different from the other drugs, occurring mainly in liver cells instead of kidneys [99]. Furthermore, the erasing of the drug from the bloodstream occurs in the free form and in the FVIII-vWF form [100]. If the drug is removed as free form, it is firstly inactivated by a process called catabolism [95]. This reaction aims to target CFC A2 domain to destabilize the protein. It may be through a spontaneous dissociation, since the A2 domain has a weakly interaction with A1/A3-C1-C2 structure, or through a proteolytic cleavage played by the activated protein C or by FXa [95].
Investigations support that the FVIII clearance is mainly by the low-density lipoprotein receptor-related protein (LRP1), an endocytic receptor expressed commonly on hepatocyte membrane and Kupffer cells and also in vasculature structures such the surface of smooth muscle cells, fibroblasts and macrophages [96, 101, 102]. The LRP1 receptor has activity towards the A3 domain (at 1804-1834), A2 domain (at 484- 509), and C-terminal of the C2 domain [95]. The first two domains present a high affinity towards LRP1 [102] whereas, the latter domain shares the same site as vWF in binding to FVIII [95], justifying why vWF reduces FVIII clearance, mediated by LRP1, at an extent of almost 90% [95]. In contrast, when both are not linked together, C2 domain may act as another site for LRP1 to exert the clearance of FVIII [101]. Nonetheless, LRP1 has been associated with polymorphisms, in particular, the LDLR c.1773C/T genotype, influencing drug pharmacokinetics and may also play a part in the inter-individual responses to treatment [103].
Cell surface heparin sulfate proteoglycans (HSPGs) are components from the extracellular matrix that act as co- receptors of LRP1 or as independent receptors (catabolic receptors) [101]. Specifically, in vivo HSPGs interacts with the A2 domain (at 558- 565) [95] and facilitates the presentation to the fragments of LRP1 [102]. Hence, it is possible to prolonged the half-life of FVIII if, simultaneously, the HSPGs and LRP1 receptors are blocked, like previously demonstrated in mice [101]. Another receptor involved in FVIII clearance is the asialoglycoprotein or Ashwell receptor (ASGPR), which is expressed by hepatocytes and structurally composed of two transmembrane protein subunits, the asialoglycoprotein receptor-1 (ASGR-1) and the asialoglycoprotein receptor-2 (ASGR-2) [97]. Its activity towards not only the unbounded FVIII, through the B-domain [104], but also the complex itself [97]. In mice, when ASGR-1 was blocked, levels of FVIII and vWF raised versus ASGR- 2 [97]. Hence, targeting ASGR-1 seems to be a good strategy to reduce the elimination process of FVIII.
More receptors Table 6 have been found in hepatic macrophages and liver sinusoidal endothelial cells (LSECs), exhibiting endocytic mechanisms against FVIII and vWF. They are currently considered the new major in vivo regulators [96]. Additionally, LSECs are the cells with the highest capacity of endocytosis as they offer high ability for lysosomal activities important to the clearance of several blood components [105]. However, their specific role in regulating the clearance of both forms of FVIII remains unclear [96]. CFCs are excreted by the liver, either as FVIII-free or FVIII-vWF, through the cellular mechanisms involving several receptors Figure 6. The role of the kidneys is negligible regarding CFCs elimination [99].
| Cell Location | Receptor | |
|---|---|---|
| Kupffer cells | LRP1 | |
| Hepatocyte's membrane | LRP1 | |
| Hepatocyte's membrane | ASGPR* | |
| Hepatocyte's membrane | ASGR-1 | |
| Hepatocyte's membrane | ASGR-2 | |
| Hepatic macrophages | HSPGs* | |
| Hepatic macrophages | LDL-R* | |
| Hepatic macrophages | SR-A1 | |
| Hepatic macrophages | LRP1 | |
| Hepatic macrophages | MGL | |
| Hepatic macrophages | Siglec-5* | |
| Hepatic macrophages | Liver sinusoidal endothelial cells | STAB2 |
| CLEC4M | Liver sinusoidal endothelial cells | |
Table 6: Summary of the potential clearance receptors involved in FVIII elimination [96,97,102]. ASGPR: Ashwell receptor; ASGR-1:
Table 6: Summary of the potential clearance receptors involved in FVIII elimination [96, 97, 102]. ASGPR: Ashwell receptor; ASGR-1: asialoglycoprotein receptor-1; ASGR-2: asialoglycoprotein receptor-2; CLEC4M: C-type lectin domain family 4-member M; HSPGs: heparin sulfate proteoglycans; LRP1: low-density lipoprotein receptor-related protein; LDL-R: low-density lipoprotein receptor; MGL: macrophage galactose-type lectin; SR-A: scavenger receptor class A member; Siglec-5: sialic acid binding immunoglobulin-like lectin 5; STAB2: stabilin-2 *in vitro binding FVIII results.
![Figure 6: The role of the kidneys is negligible regarding CFCs elimination [99].](/fulltextimages/9794/fig_6.png)
Figure 6: Representation of different receptors in liver cells. ASGPR: Ashwell receptor; CLEC4M: C-type lectin domain family 4 member M; FVIII: factor VIII; HSPGs: heparin sulfate proteoglycans; LRP1: low-density lipoprotein receptor-related protein; LDL-R: low-density lipoprotein receptor; LSECs: liver sinusoidal endothelial cells; MGL: macrophage galactose-type lectin; SR-A: scavenger receptor class A member; Siglec-5: sialic acid binding immunoglobulin-like lectin 5; STAB2: stabilin-2; t1/2: terminal half-life; vWF: von Willebrand Factor.
Pharmacokinetic Analysis of FVIII Concentrates
Usually, pharmacokinetic behavior is determined by repeated measurements of drug concentrations in plasma over the time. However, for clotting factors, “drug concentrations” differ from those of general drugs. As well- known, clotting factors are endogenous zymogens thus, their activity is measured by bioassays such as the one-stage or chromogenic assays (section 2.1) [106]. Hence, the obtained results are often understood as “plasma concentrations” which semantically is not well accepted. Instead, the terms are “activity” or “level” of FVIII in plasma [10].
Pharmacokinetics analysis regards the determination of standard and specific parameters such as follows.
Standard pharmacokinetic parameters: Standard parameters include “basic” pharmacokinetic parameters that characterize the general pharmacokinetic processes [10, 27]. For instance, the distribution is evaluated through the volume of distribution (Vd), which corresponds to the apparent volume, in which, the drug distributes to achieve the same activity levels as observed in plasma [107]. Therefore, after the infusion, Vd is achievable following the Equation1:
( ) ( ) Dose IU/kg Vd= Plasma Level IU/mL or dl Literature value for Vd of CSF is approximately 48 mL/ kg (i.e. 0.048 L/kg), which is close to the plasma volume (i.e. 3L) [108]. Nonetheless, when the FVIII concentrates infusion equals the elimination, steady-state conditions are attainable [94] and, hence, the volume at the steady-state (Vss) is more suitable, as it represents the equilibrium between compartments (plasma and surrounding tissues). The value of Vss will always exceed the Vd, suggesting that, even the large complexes like FVIII are not totally confined to the plasma space. The formula for Vss calculation is presented in Table 7 [106].
| Units | Equation | Definition | |
|---|---|---|---|
| Volume of Distribution at the steady state (Vss) | mL/kg | MRT × CL | Theoretical volume necessary for a certain amount of drug achieve the same activity level as observed in plasma, upon equilibrium between plasma and surrounding tissues. |
| Clearance (CL) | mL/h/kg | Dose/AUC | Volume of plasma cleared of drug per time unit. |
| Mean Residence Time (MRT) | h | AUCM/AUC | The average amount of time that a single molecule unit of the drug remains in plasma or body. |
| Half-life (t1/2) | h | MRT/1.443 | Time to plasma activity level decrease by ½ after equilibrium has reached. |
| Half-life (t1/2) | h | ln2/ke | Terminal half-life is a linear regression of logarithmic points in the last portion of activity portion, as elimination becomes constant. |
Table 7: Summary of the basic/fundamental pharmacokinetic parameters of FVIII concentrates [93,109]. AUC: area under the curve; A
The mean residence time (MRT) also describes the distribution of a drug [106] and it is influenced by Vss and elimination. In addition, MRT estimation (Table 7) depends on the area under of FVIII concentration vs. time curve (AUC) and the area under the first moment of the curve (AUMC) [109]. Both AUC and AUMC are extrapolated to infinity and assess by trapezoid method following Equations 2 and 3, respectively as C1 and t1, being the first plasma level and the respective time while C2 and t2 are the second measures of plasma and correspondent time [106]:
( ) ( ) C1 C2 x t2 t1 AUC 2
$$ I = \frac {\left(C _ {1} + C _ {2}\right) x \left(t _ {2} - t\right)}{2} $$
Equation 2
( ) ( ) C1 x t1+C2 x t2 x t2-t1 AUMC= 2
Equation 3
Furthermore, drug elimination is characterized by clearance (CL) as the value serves to understand the efficacy of the organs such kidneys and/or liver in removing the drug from plasma [94]. The results should be interpreted as the volume of plasma cleared of FVIII concentrates per time unit. This parameter has the particularity to be the constant of proportionality between the rate of elimination and plasma levels. Literature mean CL values in healthy adults with 70 kg surround the 200 mL/h [10] or 3 mL/h/kg [110]. Hence, the amount of FVIII excreted remains constant per unit of time [109]. As so, the constant of elimination (Ke) is assessed by Equation 4, as C1 and C2 two plasma FVIII levels within the terminal section of the curve whilst t1 and t2 are the matching time points [106].
$$ \mathrm {K e} = \frac {\left(\ln \mathrm {C} 1 - \ln \mathrm {C} 2\right)}{\left(\mathrm {t} 2 - \mathrm {t} 1\right)} $$ Equation 4 ( ) Moreover, the terminal half-life (t1/2), which is clinically useful to express the rate of the overall elimination during the terminal phase, is calculated resorting to the Equation evident that t1/2 depends on other kinetics parameters, the CL and Vd, being defined as a hybrid pharmacokinetic parameter [74].
1/2 ln2 x Volume of distribution t = Plasma clearance Equation 5
As a hybrid parameter, it is difficult to associate values of terminal half-life time to clinical features of the patients such as age, body weight or, liver diseases [74]. Nonetheless, terminal half-life time is an accurate value that can be used to define dose regimens, particularly regarding prophylaxis (Section 8.2.1). Reported average values of plasma half-life time of FVIII vary between 12-14 hours [110], sustaining the inter-patient variability [111]. Indeed, among 42 individuals with severe HA, half-life time varied between 7.4 to 20.4 hours. Furthermore, other recent studies have also reported intervals ranging from 6 to 25 hours [42] or 5.3 hours to 28.8 hours [96]. Several factors determine the pharmacokinetics as it will be in deeply discussed in section 8.4. Specific pharmacokinetic parameters: Specific parameters are a better choice to evaluate the effectiveness of regimens and to improve their safety in a specific patient [27]. They involve patient clinical events in estimation and interpretation of the results which are useful for therapeutic drug monitoring.
Descriptive specific parameters include the maximum or peak of FVIII activity (Cmax) and the minimum level, also referred as trough level (Ctrough) [27]. Furthermore, AUC and the time spent above the threshold (TAT1%) are essential to prevent the risk of bleeding as it will be exploited in section Section 8.2.1 [27]. Collins, et al. Demonstrated that TAT1% is correlated with the rate of breakthrough bleeds and hemarthrosis [112]. It is estimated by combining the administered dose and the half-life time of FVIII in a specific patient, requiring an extensive pharmacokinetic analysis that involves 5 to 6 samples collected on two or three days after drug infusion to calculate the half-life time [27].
Incremental in vivo recovery (IVR), also named in vivo recovery or simply recovery is a specific parameter that directly gives the rise (recovery) of the plasma FVIII activity after dose administration [27]. IVR corresponds to the ratio between peak level directly measured in the patient and the expected peak level (Equation 6), which is assessed either through body weight (BW) or plasma volume [106]. The average value, in adults, may vary between 0.020-0.025 IU/ dL per IU/kg [113].
( ) ( ) ( )
Observed peak IU/dL IVR IU/dL per IU/kg = Expected peak IU/dL
Equation 6
Pharmacokinetic Parameters used for Prophylaxis
Prophylaxis rationale emerged from the observation of fewer bleeding events on mild/moderate patients that have levels of FVIII ranging from 1 to 5 IU/dL. Therefore, it was straightforward hypothesized that a Ctrough above 1 IU/ dL was the ideal to reverse the severe state into a milder state [114]. Additionally, as the time spent below the Ctrough increases, higher is the risk of bleeding and breakthrough bleeds. Additionally, measuring TAT1% along with Ctrough is essential to understand the pharmacokinetic response and adjust prophylaxis regimens to the patient lifestyle [114]
Another parameter to rationally perform prophylactic regimens and increase their effectiveness is the t1/2 [115]. As studied by Collins, et al. Among children and adults that were administered with 30 IU/kg, the ones with shorter t1/2 reached the Ctrough of 1% more quickly (44 hours and 46.4 hours, respectively) than those with longer t1/2 (78 hours and 103.3 hours, respectively) [114]. Moreover, other studies demonstrated that peak levels and AUC also describe the efficacy of prophylaxis [116]. A post hoc comparison between pharmacokinetic-guided prophylaxes with standard weight adjustments prescribing ADVATE® concluded that peak levels and AUC is associated with the risk of joint and non- joint bleeding. Specifically, higher values of both parameters were positively correlated with lower bleeds. However, it is important to establish that these findings are for prophylaxis given every third day in severe patients and it cannot be extrapolated to other regimens [116].
In clinical practice, it is common to question which pharmacokinetic parameter should be used for prophylaxis. Ideally, each parameter should be tailored to the circumstances of the patient lifestyle [43]. For instance, an individual with HA that frequently practices sports is more prone to the risk of bleeding and injuries than sedentary patients [92]. Therefore, assessing the time to attain peak levels is more relevant to define the exact time of the next infusion [90, 92]. In opposition, if the patient is more sedentary, it is more important to maintain minimal FVIII levels to protect against bleedings. In case of patients who do not properly adhere to the treatment, the best scenario includes the reduction of dose frequency along with the trough level and TAT1% [27].
Pharmacokinetic Parameters for Surgeries or On-demand
In surgeries or acute bleedings, the goal is to achieve a level above the Ctrough but not higher than Cmax [42]. To manage these clinical situations, peak levels and IVR are the most important parameters to consider. Peak levels depend on the location of the bleeding and the severity of the surgery Table 8. IVR equation usually used is Equation 7 which is simplified by the ratio of the post-infusion peak level (IU/dL) and the infused dose (IU/kg) [10, 117].
| Lower-dose regimen | High-dose regimen | ||||
|---|---|---|---|---|---|
| Hemorrhage Type | Peak factor level (IU/ dL) | Duration (days) | Peak factor level (IU/dL) | Duration (days) | |
| Joint | Oct-20 | 1-2a | 40-60 | 1-2a | |
| Superficial muscle No neurovascular compromise (except lipossomas) | Oct-20 | 2-3a | 40-60 | 2-3a | |
| Iliopsoas or deep muscle with neurovascular injury OR substantial blood loss | |||||
| Initial | 20-40 | 01-2 | 80-100 | 01-Feb | |
| Maintenance | Oct-20 | 3-5b | 30-60 | 3-5b | |
| Intracranial Bleeding | |||||
| Initial | 50-80 | 01-3 | 80-100 | 01-Jul | |
| 20-40 | 8-14 | 50 | Aug-21 | ||
| Maintenance | 30-50 | 04-7 | - | - | |
| Throat and Neck | |||||
| Initial | 30-50 | 01-3 | 80-100 | 01-Jul | |
| Maintenance | Oct-20 | 04-7 | 50 | Aug-14 | |
| Gastrointestinal | |||||
| Initial | 30-50 | 01-3 | 80-100 | Jul-14 | |
| Maintenance | Oct-20 | 04-7 | 50 | ||
| Renal | 20-40 | 03-5 | 50 | 03-May | |
| Deep laceration | 20-40 | 05-7 | 50 | 05-Jul | |
| Major surgery | |||||
| Pre-operative | 60-80 | - | 80-100 | - | |
| 30-40 | 01-3 | 60-80 | 01-Mar | ||
| Post-operativec | 20-30 | 04-6 | 40-60 | 04-Jun | |
| Oct-20 | 7-14 | 30-50 | Jul-14 | ||
| Minor surgery | |||||
| Pre-operative | 40-80 | - | 50-80 | - | |
| Post-operatived | 20-50 | 01-5 | 30-80 | 01-May |
Table 8: Guidance in peak levels and duration of administration of FVIII concentrates for treatment in acute bleedings and/or sur
( ) ( ) Pos infusion peak IU/dL IVR= Dose IU/kg
Equation 7
However, the peak of the activity itself is not found directly after the end of infusion. Indeed, it is reported to be attained at, at least, 10 to 15 minutes or later (1 to 2 hours), making the IVR dependent on rigorous sampling processes [10]. These discrepancies evidence the inter- patient variability and, hence, the pharmacokinetic protocols recommend extrapolation of plasma activity based on three samples (15, 30, and 60 minutes) [27]. Others also use IVR- extrapolated at time 0 which is equivalent to the Cmax of FVIII at that time, being the ratio of Cmax and dose the right equation [117]. Regardless, it has also been reported that IVR is important to determine the loading dose of a new CFC as it only requires two samples, one at the baseline and the other after post-infusion, following Equation 8 [106]:
( ) ( ) ( ) Nº IU required=BW kg x desired rise IU/dL x Reciprocal IVR IU/kg per IU/dL Drug Cmax (IU/ dL) AUC/Dose (IU*h/dL per IU/kg)) t1/2 (h) CL MRT Vss (mL/kg) Incremental recovery (IU/dL per IU/kg) Age group (mL/h/kg) (h) STANDARD HALF-LIFE Third Generation ADVATE® Adults ≥ 18 years 111.3 ± 27.1
NR
12.9 ± 4.3
$$
3. 6 \pm
- 2 \quad \mathrm {N R} \quad \mathrm {N R} \quad
- 2 \pm
5-12 years 100.5 ± 5.6 NR 11.8 ± 3.8 3.8 ± 1.5 NR NR 2.0 ± 0.5
Equation 8
A May be longer if necessary; b Sometimes longer as secondary prophylaxis during physical therapy; c Duration referring to sequential days post-surgery (depending on treatment and the patient response); d Depending on procedure; doses will depend on half-life of treatment used.
Pharmacokinetics of FVIII concentrates
ADVATE® is the reference drug used in comparative and bioequivalence studies for new drugs. Its pharmacokinetics was studied in 195 subjects with severe HA through at all age window Table 9 while the pharmacokinetics of moroctcog alfa (ReFacto AF®) was assessed on adolescents and adults (Table 9). Direct comparisons of moroctcog alfa (ReFacto AF®) and ADVATE® were performed in 30 patients (>12 years old) to prove bioequivalence. The results of pharmacokinetics parameters (AUC0-t; AUC0-∞; incremental recovery) were all within the 80-125% interval (i.e. 90% CI) Table 10 [54].
| Children 2-5 years | 90.8 ± 19.1 | NR | 9.6 ± 1.7 | 4.8 ± 1.5 | NR | NR | 1.8 ± 0.4 |
|---|---|---|---|---|---|---|---|
| MOROCTOF ALFA | |||||||
| ≥ 12 years | NR | NR | 14.8 ± 5.6 | 2.4 ± 0.75b | 20.2 ± 7.4 | NR | 2.4 ± 0.38 |
| TUROCTOG ALFA | |||||||
| ≥ 12 years | 163 ± 50 | NR | 11.22 ± 6.86 | 2.86 ± 0.94 | 14.54 ± 5.77 | 38.18 ± 10.24 | 2.9 ± 0.6 |
| 6-12 years | 125 ± 27 | NR | 9.42 ± 1.52 | 3.70 ± 1.00 | 11.61 ± 2.32 | 41.23 ± 6.00 | 2.5 ± 0.6 |
| < 6 years | 112 ± 31 | NR | 9.99 ± 1.71 | 4.59 ± 1.73 | 12.06 ± 1.90 | 55.46 ± 23.53 | 2.2 ± 0.6 |
| OCTOGOG ALFA | |||||||
| ≥18 years | NR | NR | 14.8 ± 34 | 0.03 ± 38 | NR | 0.56 ± 14 | NR |
| 12-18 years | NR | NR | 13.3 ± 24 | 0.03 ± 27 | NR | 0.61 ± 14 | NR |
| 6-12 years | NR | NR | 14.1 ± 31 | 0.04 ± 35 | NR | 0.77 ± 15 | NR |
| < 6 years | NR | NR | 13.3 ± 24 | 0.05 ± 25 | NR | 0.92 ± 11 | NR |
| LONOCTOCOG ALFA | |||||||
| Adults ≥18 years | 106 ± 18.1 | NR | 14.2 ± 26.0 | 2.90 ± 34.4 | 20.4 ± 25.8 | 55.2 ± 20.8 | 2.00 ± 20.8 |
| Adolescents 12- 18 years | 89.7 ± 24.8 | NR | 14.3 ± 33.3 | 3.80 ± 46.9 | 20.0 ± 32.2 | 68.5 ± 29.9 | 1.69 ± 24.8 |
| Children 6-12 years | 83.5 ± 19.5 | NR | 10.2 ± 19.4 | 4.63 ± 29.5 | 12.3 ± 16.8 | 67.1 ± 22.3 | 1.66 ± 19.7 |
| < 6 years | 80.2 ± 20.6 | NR | 10.4 ± 28.7 | 5.07 ± 29.6 | 12.4 ± 25.0 | 71.0 ± 11.8 | 1.60 ± 21.1 |
| Fourth Generation | |||||||
| SIMOCTOCOG ALFA | |||||||
| ≥ 12 years | NR | NR | 14.7 ± 10.4 | 3.0 ± 1.2 | NR | NR | 2.5 ± 0.4c |
| 6-12 years | NR | NR | 10.0 ± 1.9 | 4.3 ± 1.2 | NR | NR | 1.9 ± 0.4 c |
| < 6 years | NR | NR | 9.5 ± 3.3 | 5.4 ± 2.4 | NR | NR | 1.9 ± 0.3 c |
| EXTENDED HALF-LIFE | |||||||
| PEGylation | |||||||
| RURIOCTOCOG ALFA | |||||||
| Adults ≥18 years | 145 ± 29 | NR | 15.01 ± 3.89 | 2.16 ± 0.75 | 19.70 ± 5.05 | 0.40 ± 0.09 | 2.87 ± 0.61 |
| Adolescents 12- 18 years | 117 ± 28 | NR | 13.80 ± 4.01 | 2.58 ± 0.84 | 17.73 ± 5.44 | 0.54 ± 0.22 | 2.34 ± 0.62 |
| Children 6-12 years | NR | NR | 11.93 ± 2.58 | 2.80 ± 0.67 | 17.24 ± 3.73 | 0.46 ± 0.04 | NR |
| < 6 years | NR | NR | 12.99 ± 8.75 | 3.49 ± 1.21 | 18.74 ± 12.6 | 0.54 ± 0.03 | NR |
| DAMOCTOCOG ALFA PEGOL | |||||||
| ≥ 12 years | 163 ± 14.7 | NR | 17.1 ± 27.1 | 0.016 ± 33.7d | 24.4 ± 27.5 | 0.391 ± 16.3 | NR |
| TUROCTOG ALFA PEGOL | |||||||
| ≥ 18 years | 134.4 ± 23 | NR | 19.9 ± 34 | 1.4 ± 32 | 25.2 ± 29 | 37.7 ± 27 | 2.63 ± 22 |
| 12-18 years | 133.2 ± 9 | NR | 15.8 ± 43 | 1.5 ± 43 | 21.7 ± 45 | 33.4 ± 10 | 2.79 ± 12 |
| 6-12 years | 119.6 ± 25 | NR | 14.2 ± 26 | 2.4 ± 40 | 17.3 ± 31 | 41.2 ± 25 | 1.99 ± 25 |
| < 6 years | 101.2 ± 28 | NR | 13.6 ± 20 | 2.6 ± 45 | 17.0 ± 22 | 44.2 ± 34 | 1.80 ± 29 |
| Fc Fusion | |||||||
| EFMOROCTOCOG ALFAe | |||||||
| ≥ 15 years | 131 | 47.5 | 20.9 | 2.11 | 25 | 52.6 | 2.49 |
| (104-165) | (41.6-54.2) | (18.2-23.9) | (1.85-2.41) | (22.4-27.8) | (47.4-58.3) | (2.28-2.73) | |
| 12-18 years | NR | 40.8 | 17.5 | 2.45 | 23.5 | 57.6 | 1.91 |
| (29.3-56.7) | (12.7-24.0) | (1.76-3.41) | (17.0-32.4) | (50.2-65.9) | (1.61-2.27) | ||
| 6-11 years | NR | 32.8 | 15.9 | 3.05 | 20.7 | 63.1 | 2.08 |
| (28.2-38.2) | (13.8-18.2) | (2.62-3.55) | (18.0-23.8) | (56.3-70.9) | (1.91-2.25) | ||
| < 6 years | NR | 25.9 | 14.3 | 3.86 | 17.2 | 66.5 | 1.88 |
| (23.4-28.7) | (12.6-16.2) | (3.48-4.28) | (15.4-19.3) | (59.8-73.9) | (1.73-2.05) |
Table 9: Human pharmacokinetic parameters of Factor VIII concentrates available in clinical practice. A Parameters expressed as I
Table 9: Human pharmacokinetic parameters of Factor VIII concentrates available in clinical practice. A Parameters expressed as IU*h/mL); b Parameters expressed as mL/h/kg; c Parameters expressed as percentage per IU/kg; d Parameters expressed as dL/h/kg; e Results from Efmoroctocog alfa are expressed as mean (minimal value-maximal value); NR, not reported.
| Concentrate | AUC∞ | AUCt | In vivo Recovery (%) | Incremental recovery |
|---|---|---|---|---|
| (IU*h/mL) | (IU*h/mL) | (IU/dL per IU/kg) | ||
| ReFacto AF® | 14.7 ± 6.1 | 13.8 ± 5.7 | 112 ± 22 | 2.35 ± 0.47 |
| ADVATE® | 16.5 ± 6.3 | 15.0 ± 5.4 | 114 ± 30 | 2.39 ± 0.65 |
| 90% log- transformed CI | 81.6–94.8% | 83.3–96.9% | ND | 92.5–108% |
Table 10: Pharmacokinetic parameters and bioequivalence results between ReFacto AF® and ADVATE® [54]. AUCt: area under the plasma
The pharmacokinetics parameters of Turoctocog alfa (NovoEight®) were also accessed after a single infusion of 50 IU/kg (Table 9). In addition, the bioequivalence was tested with ADVATE®, involved 23 male patients with severe Concentrate AUC (IU*h/ mL) AUCt (IU*h/mL) Cmax (IU/mL) Incremental recovery CL (mL/h/ kg) t1/2 NovoEight® 11.9942 11.3044 0.9723 0.01839 4.1687 9.8586 ADVATE® 11.8128 11.1397 0.9858 0.01816 4.2327 10.5524
Regarding octocog alfa (Kovaltry®), its pharmacokinetics was evaluated at LEOPOLD 1, on 26 previous treated patients after a single infusion of 50 IU/kg and it is presented in Table 9, demonstrating similarities with the aforementio9ned FVIII concentrates. In its turn, the last 3rd generation drug, the HA receiving the 50 IU/kg of each drug with four days of washout [118]. Accordingly, the results of pharmacokinetics endpoints were within the 90% of confidence interval (80- 125%), proving their bioequivalence Table 11.
(IU/dL per IU/kg) (h)
lonoctocog alfa (Afstyla®) demonstrated a slightly enhanced half-life time (14.5 ± 3.8h vs. 13.3 ± 4.4h) and a reduction in clearance by 28–31% in comparison to ADVATE®, probably due to the proposed technology [49, 119]. Its pharmacokinetic parameters are summarized in Table 9.
The pharmacokinetics of the 4rd generation drug (simoctocog alfa, Nuwiq®) was evaluated in 22 adolescents/ adults in GENA-01 program whilst, the estimations from children were done in GENA-03, which enrolled 26 patients. All of them received 50 IU/kg infusion of simoctocog alfa (Table 9) [63]. Overall, the drug proved to be effective in treating bleeds with standard prophylaxis (30–40 IU FVIII/ kg) both in adults and children, as well as safer in terms of adverse effects and immunologic response.
Regarding the EHL drugs, the first one to be introduced in the market was the Adynovi® (rurioctocog alfa pegol) and its pharmacokinetics (Table 9) evidenced its extended half-life time (14-19.6 hours), which is 40% longer than octocog alfa (ADVATE®) [64]. Similarly, damoctocog alfa pegol (Jivi®) exhibited a higher half-life time (Table 9, 19 hours versus 13 hours) with a median ABR between 1.9 and 3.9 depending on the prophylactic days of infusion (five and seven respectively). Furthermore, the pharmacokinetics of turoctocog alfa pegol (Table 9) evidence an extensive half-life time, ranging from 11.6h to 27.3h. This characteristic seems to contribute to the good control of bleeding by prophylaxis (median ABR of 1.3) in treated patients [7].
The only EHL drug obtained by Fc Fusion, the efmoroctocog alfa (Elocta®), revealed, after a single infusion of 50 IU/kg, a longer half-life (1.5 to 1.7-fold time) than ADVATE®, contributing for a geometric mean of half-life time of approximately 19h with single infusions versus 11-12h of ADVATE® [120]. In addition, efmoroctocog alfa prolonged the TAT1%. Particularly considering children subpopulations, the same conclusions were registered in half- life times, which were 1.4-fold (6-11 years) and 1.7-fold (<6 years) longer than in patients treated with ADVATE® (n=16) [120]. When administered as a prophylaxis regimen (25–65 IU/kg every 3–5 days), the average ABR of efmoroctocog alfa was 1.6 and the bleeding episodes (757 in total) were well manageable with 1-2 doses and a rate of success of 91.8% [64, 120].
Factors that Contribute to Inter-Individual Variability
Von willebrand factor: As already mentioned, vWF is a plasma glycoprotein that interacts with certain domains of FVIII, resulting in the vWF-FVIII complex that (a) stabilizes the FVIII structure as a heterodimer in the bloodstream facilitating the activation of thrombin; (b) protects FVIII from proteolysis cleavage by FXa or activated protein C; (c) modulates interaction with serine protease FIXa; and (d) regulates the cellular uptake within clearance circulation removal [97, 102].
Furthermore, studies have shown that vWF levels (vWF:Ag) are positively correlated with half-life time of FVIII. For each rise of 0.1 IU/dL in vWF: Ag, there is an increase of 16.6 minutes in the half-life time of FVIII. Furthermore, plasma FVIII and vWF levels can vary between 0.5 to 2 IU/ mL in healthy individuals counting for 15% of the inter- individuality [96].
ABO blood type: ABO blood group is a system of antigens consisting on three determinant structures such A, B, and H [96]. A study made in twins demonstrated that approximately 30% of plasma levels of vWF were influenced by the ABO blood group [121]. Moreover, as FVIII presents affinity towards vWF, the ABO blood group will indirectly contribute to the variability observed in FVIII pharmacokinetics and response. In fact, patients from O blood group have 20- 30% lower vWF:Ag levels [121] and, hence, as previously explained, they exhibit a shorter half-life time (15.3 h vs 19.7h) and an increased clearance [96, 122].
Although the mechanism is not yet elucidated, some hypotheses have been purposed. Before secretion, vWF is submitted to glycosylation with some ABO (H) structure similarities [96]. One is a possible effect of the ABO group on the N-linked oligosaccharide of vWF chains as they share carbohydrate structure similarities [121]. It was also studied the chance of Rhesus blood group (RhD) phenotype, defined by the protein present in erythrocytes membrane, target the FVIII pharmacokinetics but no influence on pharmacokinetics was observed [121].
Gender and race: Mean levels of vWF and FVIII were demonstrated to be significantly higher in females than males. As for ethnicities, levels of FVIII and vWF are 20% higher in African Americans than Caucasians [97, 121]. Typically, the prevalence of the ABO system tends to vary within racial groups. However, the influence in FVIII clearance remains consistent in studies that involved different ethnicities [96, 121].
Age: Age is associated with changes in several organs starting with maturation throughout the pediatric phase [123] and a decline of the functions from adults to elderly [124]. These changes are also observed in the coagulation system as aging rises several clotting factors, particularly FVIII [125].
Several studies revealed an inverse relationship between age and clearance of FVIII, which, in turn, will influence drug half-life time [126]. Clearance of FVIII showed to be greater in children than adults while, the half-life is described as shorter in children aged between 1-6 years (9.4 hours) than in patients within 10 and 65 years old (11.1 hours) [93, 96].
Younger ages have a liver ratio mass of 30-35% as this tends to decrease with aging as well as their respective functions [124]. For CFCs, this means that elimination by the liver will be at a lower rate, prolonging FVIII half-life time. Another possible explanation is the lower expression of LRP1 observed in aging rats [127]. Moreover there is also a positive correlation between aging and vWF levels [128] and, consequently, older patients present more vWF complexed with FVIII, decreasing drug clearance and increasing drug residence time [96].
Moreover, since vWF is also influenced by the ABO blood group, one study was able to analyzed the same influence but associated with aging [129]. Accordingly, 207 individuals were divided into three age categories (young, middle, or older) and it was measured vWF:Ag levels as well as the ABO antigen. The authors concluded that the ABO blood system is submitted to changes throughout ages and, A- and B-antigens were the main factors for high values of vWF, supporting the increased levels of vWF levels (0.16 IU/dL), being more significant in non-O individuals than O blood group [96].
Body weight: The increase of BW leads to higher body fat and therefore there is less plasma volume available per kg of BW [130]. Usually, patients are classified, in terms of weight, through the body mass index (BMI) determined through Equation 9, where BW is the body weight and H is patient height:
$$\text{BMI} = \frac{\text{BW}}{\text{H}^2}$$
Equation 9
An underweight patient is someone with BMI <20 kg/m² whereas, overweight has BMI >25 kg/m² [131]. Additionally, obesity is diagnosed when patients present a BMI >30 kg/m² and subdivided as moderate (BMI: 30-35 kg/m²), severe (BMI: 35-40 kg/m²), morbid (BMI >40 kg/m²) and super-morbid (BMI >50 kg/m²). Therefore, overweight/obese patients have lower plasma volume than underweight patients, and hence higher plasma volume per kg of BW [130]. For FVIII, these situations may impact its levels if the body fat and BW compositions are underestimated in clinical practice. As aforementioned, FVIII is confined to intravascular space. The vasculature represents a small fraction of body fat tissue volume (0.005 to 0.010%) so, more or less percentage of fat in this space will not have an impact on the distribution or elimination [132]. Therefore, it has been recommended the use of ideal body weight (IBW), given by the following Equation 10 for dose estimations instead of the actual BW [130]:
$$\text{IBW(kg)} = \text{H(cm)} - 100 - \left[ \frac{\text{H(cm)} - 150}{4} \right]$$
Equation 10
Moreover, for dose adjustments according to BW, IVR is usually used, following Equation 11:
$$\text{Dose} = \frac{\text{Total body weight(kg)} \times \text{desired increased in FVIII level(%)}}{\text{IVR}}$$
Body metrics can be a tool to predict pharmacokinetic parameters and subsequently helping in dosing tailoring [133]. Typically, IVR average value is 2 IU/dL per IU/kg [108] or, also common, the value of 0.5 dL/kg [133]. There is a proportional correlation between IVR and BW and, thus, as weight increases IVR will also rise [43]. A pioneering study investigated the influence of BMI on the IVR of FVIII in 201 patients [133]. They demonstrated that BMI influenced the IVR since the IVR best value was 1.60 in patients with BMI <20 kg/m², it was 2.14 for patients with BMI ranging from 20 and 30 kg/m² and 2.70 when BMI was equal to or higher than 30 kg/m². Therefore, assuming the same IVR value (2 IU/dL per IU/kg) for every patient does not take into consideration the physiological characteristics of that patient, leading to doses errors.
Moreover, a study enrolled 46 patients with different severity of HA as well as different BMI categories and created 3 sub-groups according to their fat mass index (FMI): FMI <15.0% (N=9), FMI: 15.0% - 19.9% (N=11), and FMI ≥20.0% (N=16) [134]. Mean IVR values increased from 1.74 to 1.89 and 2.35, respectively, suggesting that higher percentage of FMI is associated to higher FVIII levels. Also, patients with FMI ≥ 20.0% were over treated while patients with FMI <15% were undertreated when IVR was assumed equal to 2, corroborating the impact of individual-variability and the need for dose individualization. At the end, the authors defended that IVR should only be used as 2.0 in patients with normal BW and FMI between 15-20%.
Noteworthy, BMI was also found to be a good predictor for pharmacokinetic parameters of FVIII, comparing to other metrics such as IBW, lean body weight, adjusted body weight, or body surface area. However, these conclusions were based on a study that did not include underweight patients, or extremely body muscle mass patients, or neither anemic ones [130]. Furthermore, BMI is useful to manage patients in surgeries or acute bleedings.
Importantly, concerning dose adjustments in children, the total of BW gives a poor description of pharmacokinetics while lean body weight or body surface area can be applied [110].
Immunogenicity: It is well known that inhibitors enhance the Vss and clearance whereas the half-life time remains unchanged and IVR decreases [10, 106]. As for "non-inhibitory" antibodies, although they do not express a function directly to FVIII [72], 42 adults with severe/moderate HA and high-titer showed a decrease in half-life timed compared to patients without antibodies [75, 96].
contrast, inhibitors do not affect IVR or distribution itself [135]. The mechanism remains unclear but these antibodies account for 17% of the inter-individual variability in the half- life time and CL of FVIII [75, 96].
Liver Diseases: As the liver is the main organ for the synthesis, but also elimination, of clotting factors, hepatic diseases such as chronic and acute liver failure or cirrhosis, have an impact on drug pharmacokinetics. FVIII is the most affected by these pathological conditions as their levels are elevated [125] as well as vW, which is increased in acute failure and liver cirrhosis [136]. The underlying mechanisms are still poorly understood but they might be related to vWF or LRP1 [136]. The vWF co-exists with FVIII, and, hence, its higher levels protect FVIII from elimination, prolonging its increased levels.
Bearing in mind that FVIII is also produced by other organs such as lungs, kidneys, spleen, lungs, and brain, these organs can also influence drug plasma levels, however no studies regarding this impact are currently available [136].
Pregnancy: Overall, healthy pregnant women experience changes in their cardiovascular system with an increased risk of stroke [137]. This is directly related to hemostatic changes with the rise of most clotting factors whilst anticoagulants factors decrease as well as the fibrinolytic activity [77].
Women with HA are rare so it is more common to report them as carriers, which are classified as obligatory (certain to have the X- chromosome) or as possible (chance to have the affected gene) [138]. However, even carrying one affected chromosome, pregnant women present 50% of the normal FVIII levels [139] and, hence, a higher risk for bleeding during and after pregnancy [138].
In terms of FVIII levels, the tendency is to rise during the first half of the pregnancy [77], however not all carriers showed the same plasma activity which ranged from 5 IU/ dL to 219 IU/dL [139], highlighting the inter- and intra- individual variability and justifying the properly manage in carriers HA women to decrease their risk of bleeding.
Individualized Treatment
Individualized regimens could be a priori, if dose adjustments are made considering patient and disease characteristics (i.e. weight, age, and genetics) or, a posteriori after the administration of the concentrate and quantification of its correspondent levels. The need of monitoring and tailoring treatment in FVIII concentrates is based on the significant inter-individual pharmacokinetic variability and on the correlation between the levels and pharmacological response of the drug [115].
Hemophilia population is considered heterogenic not only because of the distinct responses to the treatment but also because of the variability related to the bleedings [140]. As explained in section 1.1., disease severity is established and accepted by the current guidelines as an association between bleeding characteristics and residual FVIII: C. However, studies are evidencing that patients with the same residual FVIII:C can present different clinical bleedings. For example, in 5 to 10% of severe patients with levels FVIII <1IU/dL, mild bleeding phenotype was observed whereas 15% of moderate patients presented a frequent bleeding registry [111]. In terms of frequency of bleedings, it is expected that severe patients exhibit an average of 15 to 35 spontaneous joint and muscle bleeds [29]. But, similarly to the previous example, clinical irreversible arthropathy has been observed within mild and moderate patients [140]. Furthermore, this population revealed that arthropathy can happen either in patients that bleed frequently or in absence of it as a subclinical characteristic.
Besides the different clinical phenotypes, as the goal of prophylaxis is to maintain levels above 1IU/dL to prevent clinical features, assuming this limit for all HA patients may not be the ideal in clinical practice [140]. In fact, some patients with levels above 1% may not bleed whereas others may bleed with 3 IU/dL trough levels. All these arguments prove the existence of heterogenicity between HA patients [111], and, hence, monitoring every patient is important, regardless of the severity classification [140]. Furthermore, individual trough levels and TAT1% should not be considered 1 IU/dL for all of them; instead, it must be herein emphasized the dynamics of parameters that may vary from 2 IU/dL to 15-20 IU/dL to the upper limit, which is around 60-160 IU/dL [117].
FVIII concentrates have an extensive therapeutic range when compared to general drugs that are submitted to TDM. However, the risk of being sub-therapeutic is too dangerous [115]. On other hand, FVIII levels above the maximum desirable (supratherapeutic levels) can also be a risk for thrombosis even though this is less harmful and can easily be manageable.
Population pharmacokinetics (popPK) has been reliably leveraged to generate individual pharmacokinetic in hemophilia patients. Specific popPK models, recently reviewed by Goedhart, [141] are suited to predict individual pharmacokinetic under a variety of scenarios that may not be captured by clinical trials, allowing for individualized prophylactic treatment.
Conclusions
Monitoring the levels of FVIII concentrates and personalizing their regimens based on the needs of each patient can bring through a cost-effective system a lower risk of therapeutic failure and of developing adverse effects. Moreover, drug adherence is expected to increase as well as the health system costs. Importantly, patients with antibodies have higher risk of being sub therapeutic and are associated to higher health costs. Pharmacokinetics will not be just effective and safe but also well economically balanced.
It became herein evident that there is considerable inter- individual variability in the pharmacokinetics and bleeding response to FVIII concentrates that make BW-adjusted dosing far away from optimal to prevent the occurrence of bleeds but, simultaneously, guaranteeing drug therapeutic effects.
Conflicts of Interest: The authors have no conflicts of interest.
References
-
Gale AJ (2011) Continuing education course #2: current understanding of hemostasis. Toxicol Pathol 39(1): 273- 280.
-
Franchini M, Mannucci PM (2014) The history of hemophilia. Semin Thromb Hemost 40(5): 571-576.
-
Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M (2020) WFH Guidelines for the Management of Hemophilia. In: Mehta p, et al. (Eds.), Haemophilia. 3rd(Edn.), Wiley Online Library, 26 (Suppl 6): 1-158.
-
Lippi G, Franchini M, Montagnana M, Favaloro EJ (2012) Inherited disorders of blood coagulation. Ann Med 44(5): 405-418.
-
O’Donnell JS, O’Sullivan JM, Preston RJS (2019) Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br J Haematol 186(1): 24- 36.
-
Berntorp E, Fischer K (2021) Haemophilia. Nat Rev Dis Primers 7(1): 45.
-
Raso S, Hermans C (2018) Recombinant factor VIII: past, present and future of treatment of hemophilia A. Drugs Today (Barc) 54(4): 269-281.
-
Fang H, Wang L, Wang H (2007) The protein structure and effect of factor VIII. Thromb Res 119(1): 1-13.
-
World-Federation-Hemofilia (2020) Report on the Annual Global Survey 2019.
-
Bjorkman S, Berntorp E (2001) Pharmacokinetics of coagulation factors: clinical relevance for patients with haemophilia. Clin Pharmacokinet 40(11): 815-832.
-
Fijnvandraat K, Cnossen MH, Leebeek FWG, Peters M (2012) Diagnosis and management of haemophilia. BMJ 344: e2707.
-
Benson G, Auerswald G, Dolan G, Duffy A, Hermans C, et al. (2018) Diagnosis and care of patients with mild haemophilia: practical recommendations for clinical management. Blood Transfus 16(6): 535-544.
-
Peyvandi F, Kenet G, Pekrul I, Pruthi RK, Ramge P, et al. (2020) Laboratory testing in hemophilia: Impact of factor and non-factor replacement therapy on coagulation assays. J Thromb Haemost 18(6): 1242-1255.
-
Thompson AR (2003) Structure and function of the factor VIII gene and protein. Semin Thromb Hemost 29(1): 11-22.
-
Maggs PHB, Pasi KJ (2003) Haemophilias A and B. Lancet 361(9371): 1801-1809.
-
Fay PJ (2004) Activation of factor VIII and mechanisms of cofactor action. Blood Rev18(1): 1-15.
-
Carcao M, Moorehead P, Lillicrap D (2018) Hemophilia A and B. In: Hoffman R (Ed.), Hematology: Basic Principles and Practice. Elsevier Science Inc, Philadelphia, pp: 2001-2022.
-
Palta S, Saroa R, A Palta (2014) Overview of the coagulation system. Indian J Anaesth 58(5): 515-523.
-
Kumar R, Carcao M (2013) Inherited abnormalities of coagulation: hemophilia, von Willebrand disease, and beyond. Pediatr Clin North Am 60(6): 1419-1441.
-
Pisarek MA, Płucienniczak G, Ciach T, Płucienniczak A (2016)The factor VIII protein and its function. Acta Biochim Pol 63(1): 11-16.
-
Hall JE, Hall ME (2016) Hemostasis and blood coagulation. In: Hall JE (Ed.), Guyton and Hall Textbook of Medical Physiology. 14th(Edn.), Elsevier Inc, Philadelphia, pp: 1150.
-
Winter WE, Flax SD, Harris NS (2017) Coagulation Testing in the Core Laboratory. Lab Med 48(4): 295-313.
-
Peyvandi F, Kenet G, Pekrul I, Pruthi RK, Ramge P, et al. (2020) Laboratory testing in hemophilia: Impact of factor and non-factor replacement therapy on coagulation assays. J Thromb Haemost 18(6): 1242-1255.
-
Potgieter JJ, Damgaard M, Hillarp A (2015) One-stage vs. chromogenic assays in haemophilia A. Eur J Haematol 94 Suppl 77: 38-44.
-
Kitchen S, McCraw A, Echenagucia M (2010) Factor Assays Based on APTT (One- Stage Assay of FVIII:C, FIX, FXI, or FXII). In: Kitchen S, et al (Eds.), Diagnosis of hemophilia and other bleeding disorders. 2nd(Edn.), World Federation of Hemophilia, Montreal, Canada, pp: 67-69.
-
Teichman J, Chaudhry HR, Sholzberg M (2018) Novel assays in the coagulation laboratory: a clinical and laboratory perspective. Transfus Apher Sci 57(4): 480- 484.
-
Delavenne X, Dargaud Y (2020) Pharmacokinetics for haemophilia treaters: Meaning of PK parameters, interpretation pitfalls, and use in the clinic. Thromb Res 192: 52-60.
-
Marchesini E, Morfini M (2021) Recent Advances in the Treatment of Hemophilia: A Review. Biologics 15: 221- 235.
-
Jayandharan GR, Srivastava A (2008) The phenotypic heterogeneity of severe hemophilia. Semin Thromb Hemost 34(1): 128-141.
-
Melchiorre D, Manetti M, Cerinic MM (2017) Pathophysiology of Hemophilic Arthropathy. J Clin Med 6(7): 63.
-
Franchini M, Mannucci PM (2017) Modifiers of clinical phenotype in severe congenital hemophilia. Thromb Res 156: 60-64.
-
Morfini M, Rapisarda CAP (2019) Safety of recombinant coagulation factors in treating hemophilia. Expert Opin Drug Saf 18(2): 75-85.
-
Castaman G, Linari S (2018) Pharmacokinetic drug evaluation of recombinant factor VIII for the treatment of hemophilia A. Expert Opin Drug Metab Toxicol 14(2): 143-151.
-
Jenkins PV, Rawley O, Smith OP, O’Donnell JS (2012) Elevated factor VIII levels and risk of venous thrombosis. Br J Haematol 157(6): 653-663.
-
Rietveld IM, Schreuder M, Reitsma PH, Bos MHA (2018) Elevated coagulation factor levels affect the tissue factor-threshold in thrombin generation. Thromb Res 172: 104-109.
-
Yap ES, Timp JF, Flinterman LE, van Hylckama Vlieg A, Rosendaal FR, et al. (2015) Elevated levels of factor VIII and subsequent risk of all-cause mortality: results from the MEGA follow-up study. J Thromb Haemost 13(10): 1833-1842.
-
Davari M, Gharibnaseri Z, Ravanbod R, Sadeghi A (2019) Health status and quality of life in patients with severe hemophilia A: A cross-sectional survey. Hematol Rep 11(2): 7894.
-
Peyvandi F, Garagiola I, Young G (2016) The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet 388(10040): 187-197.
-
Swiech K, Picanço-Castro V, Covas DT (2017) Production of recombinant coagulation factors: Are humans the best host cells? Bioengineered 8(5): 462-470.
-
Franchini M (2013) The modern treatment of haemophilia: a narrative review. Blood Transfus 11(2): 178-182.
-
Steen Carlsson K, Höjgård S, Glomstein A, Lethagen S, Schulman S, et al. (2003) On-demand vs. prophylactic treatment for severe haemophilia in Norway and Sweden: differences in treatment characteristics and outcome. Haemophilia 9(5): 555-566.
-
Hazendonk HCAM, van Moort I, Mathôt RAA, Fijnvandraat K, Leebeek FWG, et al. (2018) Setting the stage for individualized therapy in hemophilia: What role can pharmacokinetics play? Blood Rev 32(4): 265-271.
-
Collins PW, Fischer K, Morfini M, Blanchette VS, Björkman S, et al. (2011) Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia 17(1): 2-10.
-
Astermark J, Hart D, Lobet S, Blatný J, d’Oiron R, et al. (2016) Partnering to change the world for people with haemophilia: 6(th) Haemophilia Global Summit, Prague, Czech Republic, 24-26(th) September 2015. Eur J Haematol 97(84): 3-23.
-
Blanchette VS (2010) Prophylaxis in the haemophilia population. Haemophilia 16(5): 181-188.
-
Burnouf T (2018) An overview of plasma fractionation. Annals of Blood 3.
-
Klamroth R, Gröner A, Simon TL (2014) Pathogen inactivation and removal methods for plasma-derived clotting factor concentrates. Transfusion 54(5): 1406- 1417.
-
(2004) WHO, Guidelines on viral inactivation and removal procedures intended to assure the viral safety of human blood plasma products.
-
Raso S, Hermans C (2018) Lonoctocog alfa (rVIII- SingleChain) for the treatment of haemophilia A. Expert Opin Biol Ther 18(1): 87-94.
-
Saenko EL, Ananyeva NM, Shima M, Hauser CA, Pipe SW, et al. (2003) The future of recombinant coagulation factors. J Thromb Haemost 1(5): 922-930.
-
Franchini M, Lippi G (2010) Recombinant factor VIII concentrates. Semin Thromb Hemost 36(5): 493-497.
-
Shapiro AD (2007) Anti-hemophilic factor (recombinant), plasma/albumin-free method (octocog- alpha; ADVATE®) in the management of hemophilia A. Vasc Health Risk Manag 3(5): 555-565.
-
Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG, Vorobiev AI, et al. (2013) Blood Clotting Factor VIII: From Evolution to Therapy. Acta Naturae 5(2): 19-39.
-
Recht M, Nemes L, Matysiak M, Manco-Johnson M, Lusher J, et al. (2009) Clinical evaluation of moroctocog alfa (AF- CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full-length recombinant factor VIII. Haemophilia 15(4): 869-880.
-
Ezban M, Vad K, Kjalke M (2014) Turoctocog alfa (NovoEight(R))--from design to clinical proof of concept. Eur J Haematol 93(5): 369-376.
-
Ahmadian H, Hansen EB, Faber JH, Sejergaard L, Karlsson J, et al. (2016) Molecular design and downstream processing of turoctocog alfa (NovoEight), a B-domain truncated factor VIII molecule. Blood Coagul Fibrinolysis 27(5): 568-575.
-
Lentz SR, Misgav M, Ozelo M, Salek SZ, Veljkovic D, et al. (2013) Results from a large multinational clinical trial (guardian1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia 19(5): 691-697.
-
Mahlangu JN, Ahuja SP, Windyga J, Church N, Shah A, et al. (2018) BAY 81-8973, a full-length recombinant factor VIII for the treatment of hemophilia A: product review. Ther Adv Hematol 9(7): 191-205.
-
Al-Salama ZT, Scott LJ (2017) Lonoctocog Alfa: A Review in Haemophilia A. Drugs 77(15): 1677-1686.
-
Zollner S, Raquet E, Claar P, Müller-Cohrs J, Metzner HJ, et al. (2014) Non-clinical pharmacokinetics and pharmacodynamics of rVIII-SingleChain, a novel recombinant single-chain factor VIII. Thromb Res 134(1): 125-131.
-
Schmidbauer S, Witzel R, Robbel L, Sebastian P, Grammel N, et al. (2015) Physicochemical characterisation of rVIII-SingleChain, a novel recombinant single-chain factor VIII. Thromb Res 136(2): 388-395.
-
Schiavoni M, Napolitano M, Giuffrida G, Coluccia A, Siragusa S, et al. (2019) Status of Recombinant Factor VIII Concentrate Treatment for Hemophilia a in Italy: Characteristics and Clinical Benefits. Front Med (Lausanne) 6: 261.
-
Lissitchkov T, Klukowska A, Pasi J, Kessler CM, Klamroth R, et al. (2019) Efficacy and safety of simoctocog alfa (Nuwiq(R)) in patients with severe hemophilia A: a review of clinical trial data from the GENA program. Ther Adv Hematol 10.
-
Cafuir LA, Kempton CL (2017) Current and emerging factor VIII replacement products for hemophilia A. Ther Adv Hematol 8(10): 303-313.
-
Mannucci PM (2020) Benefits and limitations of extended plasma half-life factor VIII products in hemophilia A. Expert Opin Investig Drugs 29(3): 303-309.
-
Ar MC, Balkan C, Kavaklı K (2019) Extended Half-Life Coagulation Factors: A New Era in the Management of Hemophilia Patients. Turk J Haematol 36(3): 141-154.
-
Fogarty PF (2011) Biological rationale for new drugs in the bleeding disorders pipeline. Hematology Am Soc Hematol Educ Program 2011: 397-404.
-
Mancuso ME, Mannucci PM (2014) Fc-fusion technology and recombinant FVIII and FIX in the management of the hemophilias. Drug Des Devel Ther 8: 365-371.
-
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7(9): 715-725.
-
Nakar C, Shapiro A (2019) Hemophilia A with inhibitor: Immune tolerance induction (ITI) in the mirror of time. Transfus Apher Sci 58(5): 578-589.
-
Witmer C, Young G (2013) Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol 4(1): 59-72.
-
Miller CH (2018) Laboratory testing for factor VIII and IX inhibitors in haemophilia: A review. Haemophilia 24(2): 186-197.
-
Lebreton A, Lapalud P, Chambost H, Biron-Andréani C, Morange PE, et al. (2011) Prevalence and epitope specificity of non-neutralising antibodies in a large cohort of haemophilia A patients without inhibitors. Thromb Haemost 105(6): 954-961.
-
Toutain PL, Bousquet-Mélou A (2004) Plasma terminal half-life. J Vet Pharmacol Ther 27(6): 427-439.
-
Abdi A, Bordbar MR, Hassan S, Rosendaal FR, van der Bom JG, et al. (2020) Prevalence and Incidence of Non- neutralizing Antibodies in Congenital Hemophilia A- A Systematic Review and Meta-Analysis. Front Immunol 11: 563.
-
Cugno M, Gualtierotti R, Tedeschi A, Meroni PL (2014) Autoantibodies to coagulation factors: from pathophysiology to diagnosis and therapy. Autoimmun Rev 13(1): 40-48.
-
Franchini M (2006) Haemostasis and pregnancy. Thromb Haemost 95(3): 401-413.
-
Carcao M, Goudemand J (2018) Inhibitors In Hemophilia: A Primer. In: Christine A Lee MA, et al. (Eds.), Treatment Iof Hemophilia. 5th (Edn.), World-Federation-Hemofilia, Canada 7.
-
Garagiola I, Palla R, Peyvandi F (2018) Risk factors for inhibitor development in severe hemophilia a. Thromb Res 168: 20-27.
-
Tieu P, Chan A, Matino D (2020) Molecular Mechanisms of Inhibitor Development in Hemophilia. Mediterr J Hematol Infect Dis 12(1): e2020001.
-
Cannavò A, Valsecchi C, Garagiola I, Palla R, Mannucci PM, et al. (2017) Nonneutralizing antibodies against factor VIII and risk of inhibitor development in severe hemophilia A. Blood 129(10): 1245-1250.
-
Mannucci PM, Franchini M (2017) Porcine recombinant factor VIII: an additional weapon to handle anti-factor VIII antibodies. Blood Transfus 15(4): 365-368.
-
Loomans JI, Kruip MJHA, Carcao M, Jackson S, van Velzen AS, et al. (2018) Desmopressin in moderate hemophilia A patients: a treatment worth considering. Haematologica 103(3): 550-557.
-
Tjønnfjord GE, Holme PA (2007) Factor eight inhibitor bypass activity (FEIBA) in the management of bleeds in hemophilia patients with high-titer inhibitors. Vasc Health Risk Manag 3(4): 527-531.
-
Hedner U (2006) Mechanism of action of recombinant activated factor VII: an update. Semin Hematol 43(1 Suppl 1): S105-107.
-
Blair HA (2019) Emicizumab: A Review in Haemophilia A. Drugs 79(15): 1697-1707.
-
Sankar AD, Weyand AC, Pipe SW (2019) The evolution of recombinant factor replacement for hemophilia. Transfus Apher Sci 58(5): 596-600.
-
Coppola A, Marrone E, Conca P , Cimino E , Mormile R, et al. (2016) Safety of Switching Factor VIII Products in the Era of Evolving Concentrates: Myths and Facts. Semin Thromb Hemost 42(5): 563-576.
-
Yu JK, Iorio A, Edginton AN, WAPPS co‐investigators (2019) Using pharmacokinetics for tailoring prophylaxis in people with hemophilia switching between clotting factor products: A scoping review. Res Pract Thromb Haemost 3(3): 528-541.
-
Escobar M, Santagostino E, Mancuso ME, Coppens M, Balasa V, et al. (2019)S witching patients in the age of long-acting recombinant products? Expert Rev Hematol 12(sup1): 1-13.
-
Rayment R, Chalmers E, Forsyth K, Gooding R, Kelly AM, et al. (2020) Guidelines on the use of prophylactic factor replacement for children and adults with Haemophilia A and B. Br J Haematol 190(5): 684-695.
-
Morfini M, Farrugia A (2019) Pharmacokinetic and safety considerations when switching from standard to extended half-life clotting factor concentrates in hemophilia. Expert Rev Hematol 12(10): 883-892.
-
Hermans C, Dolan G (2020) Pharmacokinetics in routine haemophilia clinical practice: rationale and modalities-a practical review. Ther Adv Hematol 11: 2040620720966888.
-
Rosenbaum E (2017) Basic Pharmacokinetics and Pharmacodynamics, an Integrated Textbook and Computer Simulations. In: Rosenbaum SE (Eds.), Basic Pharmacokinetics and Pharmacodynamics. 2nd(Edn.), JohnWiley & Sons, Inc: New Jersey, pp: 71-98.
-
Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG, Vorobiev AI (2013) Blood clotting factor VIII: From evolution to therapy. Acta Naturae 5(2): 19-39.
-
Turecek PL, Johnsen JM, Pipe SW, O’Donnell JS, iPATH study group (2020) Biological mechanisms underlying inter-individual variation in factor VIII clearance in haemophilia. Haemophilia 26(4): 575-583.
-
Terraube V, O’Donnell JS, Jenkins PV (2010) Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 16(1): 3-13.
-
Lillicrap D (2008) Extending half-life in coagulation factors: where do we stand? Thromb Res 122 (Suppl 4): S2-8.
-
Pipe SW (2010) Go long! A touchdown for factor VIII? Blood 116(2): 153-154.
-
Swystun LL, Lai JD, Notley C, Georgescu I, Paine AS, et al. (2018) The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J Clin Invest 128(9): 4057-4073.
-
Sarafanov AG, Ananyeva NM, Shima M, Saenko EL (2001) Cell surface heparan sulfate proteoglycans participate in factor VIII catabolism mediated by low density lipoprotein receptor-related protein. J Biol Chem 276(15): 11970-11979.
-
Lenting PJ, VAN Schooten CJM, Denis CV (2007) Clearance mechanisms of von Willebrand factor and factor VIII. J Thromb Haemost 5(7): 1353-1360.
-
Lunghi B, Bernardi F, Martinelli N, Frusconi S, Branchini A, et al. (2019) Functional polymorphisms in the LDLR and pharmacokinetics of Factor VIII concentrates. J Thromb Haemost 17(8): 1288-1296.
-
Mei B, Pan C, Jiang H, Tjandra H, Strauss J, et al. (2010) Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood 116(2): 270-279.
-
Poisson J, Lemoinne S, Boulanger C, Durand F, Moreauet R, et al. (2017) Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol 66(1): 212-227.
-
Shapiro AD, Korth-Bradley J, Poon MC (2005) Use of pharmacokinetics in the coagulation factor treatment of patients with haemophilia. Haemophilia 11(6): 571-582.
-
Iorio A, Blanchette V, Blatny J, Collins P, Fischer K, et al. (2017) Estimating and interpreting the pharmacokinetic profiles of individual patients with hemophilia A or B using a population pharmacokinetic approach: communication from the SSC of the ISTH. J Thromb Haemost 15(12): 2461-2465.
-
McEneny-King A, Chelle P, Henrard S, Hermans C, Iorio A, et al. (2017)Modeling of Body Weight Metrics for Effective and Cost-Efficient Conventional Factor VIII Dosing in Hemophilia A Prophylaxis. Pharmaceutics 9(4): 47.
-
Morfini M (2002)Comparative pharmacokinetic studies in haemophilia. Haemophilia 8 (Suppl 2): 30-33.
-
Bjorkman S, Folkesson A, Jonsson S (2009) Pharmacokinetics and dose requirements of factor VIII over the age range 3-74 years: a population analysis based on 50 patients with long-term prophylactic treatment for haemophilia A. Eur J Clin Pharmacol 65(10): 989-998.
-
Nogami K, Shima M (2015) Phenotypic heterogeneity of hemostasis in severe hemophilia. Semin Thromb Hemost 41(8): 826-831.
-
Collins PW, Blanchette VS, Fischer K, Björkman S, Oh M, et al. (2009) Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost 7(3): 413-420.
-
Barnes C (2013) Importance of Pharmacokinetics in the Management of Hemophilia. Pediatric Blood Cancer 60(1): S27-S29.
-
Collins PW, Björkman S, Fischer K, Blanchette V, Oh M, et al. (2010) Factor VIII requirement to maintain a target plasma level in the prophylactic treatment of severe hemophilia A: influences of variance in pharmacokinetics and treatment regimens. Journal of Thrombosis and Haemostasis 8(2): 269-275..
-
Carcao MD, Iorio A (2015) Individualizing factor replacement therapy in severe hemophilia. Semin Thromb Hemost 41(8): 864-871.
-
Valentino LA, Pipe SW, Collins PW, Blanchette VS, Berntorp E, et al. (2016) Association of peak factor VIII levels and area under the curve with bleeding in patients with haemophilia A on every third day pharmacokinetic- guided prophylaxis. Haemophilia 22(4): 514-520.
-
Morfini M (2017) The History of Clotting Factor Concentrates Pharmacokinetics. Journal of Clinical Medicine 6(3): 35.
-
Martinowitz U, Bjerre J, Brand B, Klamroth R, Misgav M, et al. (2011) Bioequivalence between two serum-free recombinant factor VIII preparations (N8 and ADVATE(R))--an open-label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia 17(6): 854-849.
-
Klamroth R, Simpson M, Depka-Prondzinski MV, Gill JC, Morfini M, et al. (2016) Comparative pharmacokinetics of rVIII-SingleChain and octocog alfa (Advate(®) ) in patients with severe haemophilia A. Haemophilia 22(5): 730-738.
-
Frampton JE (2016) Efmoroctocog Alfa: A Review in Haemophilia A. Drugs 76(13): 1281-1291.
-
Wang Z, Dou M, Du X, Ma L, Sun P, et al. (2017) Influences of ABO blood group, age and gender on plasma coagulation factor VIII, fibrinogen, von Willebrand factor and ADAMTS13 levels in a Chinese population. PeerJ 5: e3156.
-
Kamphuisen PW, Eikenboom JC, Bertina RM (2001) Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 21(5): 731-738.
-
van den Anker J, Reed MD, Allegaert K, Kearns GL (2018) Developmental Changes in Pharmacokinetics and Pharmacodynamics. J Clin Pharmacol 58(Suppl 10): S10-S25.
-
Thürmann PA (2020) Pharmacodynamics and pharmacokinetics in older adults. Curr Opin Anaesthesiol 33(1): 109-113.
-
Miesbach W, Alesci S, Krekeler S, Seifried E (2009) Age-dependent increase of FVIII:C in mild haemophilia A. Haemophilia 15(5): 1022-1026.
-
Björkman S (2013) Comparative pharmacokinetics of factor VIII and recombinant factor IX: for which coagulation factors should half-life change with age? Haemophilia 19(6): 882-886.
-
Sagare AP, Deane R, Zlokovic BV (2012) Low-density lipoprotein receptor-related protein 1: a physiological Abeta homeostatic mechanism with multiple therapeutic opportunities. Pharmacol Ther 136(1): 94-105.
-
Favaloro EJ, Franchini M, Lippi G (2014) Aging hemostasis: changes to laboratory markers of hemostasis as we age - a narrative review. Semin Thromb Hemost 40(6): 621-633.
-
Albanez S, Ogiwara K, Michels A, Hopman W, Grabell J, et al. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J Thromb Haemost 14(5): 953-963.
-
Tiede A, Rosa Cid A, Goldmann G, Jiménez-Yuste V, Pluta M, et al. (2020) Body Mass Index Best Predicts Recovery of Recombinant Factor VIII in Underweight to Obese Patients with Severe Haemophilia A. Thromb Haemost 120(2): 277-288.
-
Hunt BJ (2018) Hemostasis at Extremes of Body Weight. Semin Thromb Hemost 44(7): 632-639.
-
McEneny-King A, Iorio A, Foster G, Edginton AN (2016) The use of pharmacokinetics in dose individualization of factor VIII in the treatment of hemophilia A. Expert Opin Drug Metab Toxicol 12(11): 1313-1321.
-
Henrard S, Speybroeck N, Hermans C (2013) Impact of being underweight or overweight on factor VIII dosing in hemophilia A patients. Haematologica 98(9): 1481- 1486.
-
Henrard S, Speybroeck N, Hermans C (2011) Body weight and fat mass index as strong predictors of factor VIII in vivo recovery in adults with hemophilia A. J Thromb Haemost 9(9): 1784-1790.
-
Bjorkman S (2003) Prophylactic dosing of factor VIII and factor IX from a clinical pharmacokinetic perspective. Haemophilia 9 (Suppl 1): 109-110.
-
Hollestelle MJ, Geertzen GMH, Straatsburg IH, van Gulik TM, van Mourik JA (2004) Factor VIII expression in liver disease. Thromb Haemost 91(2): 267-275.
-
Feghali MR, Venkataramanan R, Caritis S (2015) Venkataramanan, and S. Caritis, Pharmacokinetics of drugs in pregnancy. Semin Perinatol 39(7): 512-519.
-
Leebeek FWG, Duvekot J, Kruip MJHA (2020) How I manage pregnancy in carriers of hemophilia and patients with von Willebrand disease. Blood 136(19): 2143-2150.
-
Chi C, Lee CA, Shiltagh N, Khan A, Pollard D, et al. (2008) Pregnancy in carriers of haemophilia. Haemophilia 14(1): 56-64.
-
Sorensen B, Auerswald G, Benson G, Elezović I, Felder M, et al. (2015) Rationale for individualizing haemophilia care. Blood Coagul Fibrinolysis 26(8): 849- 857.
-
Goedhart TMHJ, Bukkems LH, Zwaan CM, Mathôt RAA, Cnossen MH, et al. (2021) Population pharmacokinetic modeling of factor concentrates in hemophilia: an overview and evaluation of best practice 5(20): 4314-4325.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review