Cyto-Architectural Complexity of Dyshormonogenic Goitre can Closely Mimic a Thyroid Follicular Malignancy: A Case Report and Review of the Literature to Avoid Diagnostic Pitfall
A 13-year-old male child presented in the OPD with a midline 5x4cm thyroid swelling of 2 years duration with the chief complaint of difficulty in breathing. He also had growth and mental retardation since 3 years of age. His thyroid profile records revealed a severe degree of hypothyroidism and he was on thyroxine supplement since then. He underwent a right hemithyroidectomy for the right-sided nodular goitre causing on-and-off dyspnea. The gross specimen measured 5x4x3cm, which on cut section showed partially encapsulated multi-nodular solid areas. Microscopy yielded marked architectural complexity and prominent cellular and nuclear pleomorphism that mimicked a thyroid malignancy; for eg., a poorly differentiated papillary or follicular carcinoma. However, a thorough assessment of a range of histo-morphological features in the context of clinical history clinched the diagnosis of dyshormonogenetic goitre. One year postoperatively the patient is doing well on Thyroxine supplement. This case report reviews the literature about this rare benign entity of dyshormonogenetic goitre which closely mimics thyroid malignancy, notably a follicular malignancy on microscopy owing to its cytoarchitectural complexity and nuclear atypia. Hence, knowledge of its clinical presentation, gross and diverse microscopic features is extremely important to avoid misdiagnosis and thereby clinical management.
Introduction
Dyshormonogenetic goitre (DHG) is genetically determined familial thyroid hyperplasias due to enzyme defects in thyroid-hormone synthesis resulting in thyroid hormone insufficiency [1]. Dyshormonogenetic goitre was first described by Pendred in 1896 and Osler in 1897 [2].
This entity was previously known as congenital/familial goitre [2]. The prevalence of dyshormonogenetic goitre is a rare entity accounting for 1 per 30,000-50,000 live births [1, 2], and is the second most frequent cause of permanent congenital hypothyroidism, following thyroid dysgenesis [3]. In India, dyshormonogenesis is more common than thyroid dysgenesis [4].
It is a rare inherited disorder, resulting from a mutation in one of the seven genes encoding thyroid hormone synthesis: NIS/SLC5A5, PDS/SLC26A4, TG, TPO, DUOX2, DUOXA2 and YID/DEHALI. The two most frequent genes involved are Thyroid peroxidase and Thyroglobulin. Its higher rates are seen in consanguineous marriages and are more commonly seen in females, with a M:F ratio of 1:2. Mode of inheritance in thyroid dyshormonogenesis is autosomal recessive, with the exception of DUOX2 mutation, which may be autosomal dominant. Generally, it involves both the lobes of thyroid. The clinical presentation depends on the severity of the defect. In cases of severe defect, the patient presents in neonatal period [4]. Clinically the patient presents with diffuse enlargement of bilateral thyroid lobes with features of hypothyroidism associated with growth and intellectual disability. Patients with mild defects present in puberty or in adult life with subclinical hypothyroidism or euthyroidism. [5] DHG leads to congenital hypothyroidism, since it is one of the most treatable causes of cognitive impairment in children [4], knowing the detailed histopathological features of DHG is equally important to avoid diagnostic pitfalls.
Case Report
A 13-year-old male presented with a complaint of swelling in front of his neck since 2 years and growth retardation since 3 years of age along with intermittent difficulty in breathing for 1 year. He was from a non-endemic area, and there was no history of previous neck irradiation, consanguineous marriage, maternal history of hypothyroidism or intake of goitrogenic substances during the pregnancy. No relevant family history was present. On examination, he had a short stature for his age with signs of hypothyroidism and mental retardation. He also had macroglossia, dry, coarse skin and signs of myxoedema.
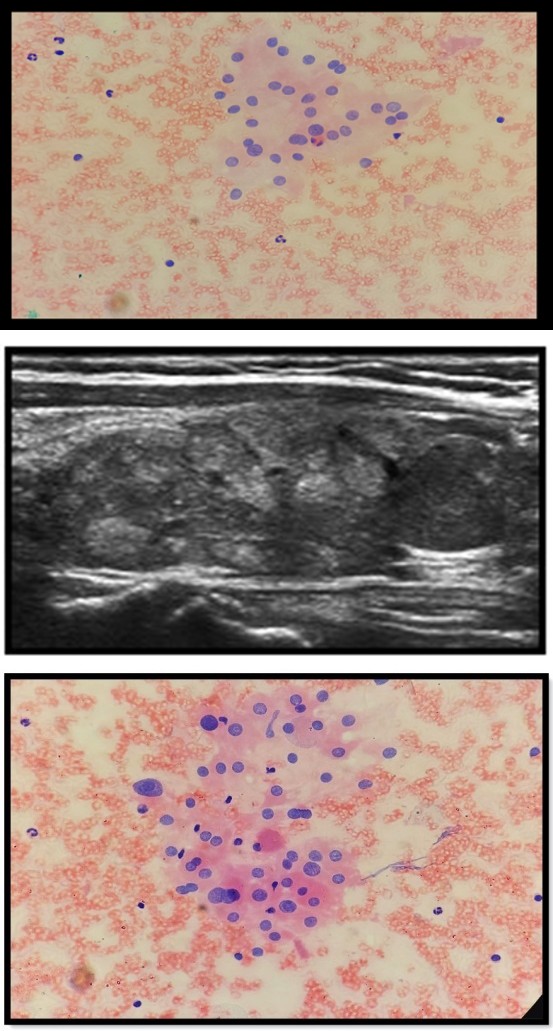
Clinically, a solitary nodular swelling of 5x4cm was present more on the right side of neck extending 1cm above the sternum and 2cm below the thyroid cartilage that moved with deglutition. There was no cervical lymphadenopathy, bruit, or retrosternal extension. A thyroid profile was advised during the course of the disease which revealed a high TSH level ranging between 15-16 IU/l and normal T3 & T4 levels. Ultrasonography neck showed a well-circumscribed multi- nodular enlargement of the right thyroid lobe with a smooth margin and internal vascularity, suggestive of multi-nodular goitre (Figure 1). No lymphadenopathy was detected.
Well circumscibed multinodular enlargement of the right thyroid lobe with smooth margins. Fine needle aspiration of the thyroid from multiple sites showed a cellular aspirate with follicular cells arranged in the micro-follicular and follicular pattern. Some follicular cells showed mild to moderate nuclear atypia, though no intranuclear inclusion or grooving was seen. The background showed scant colloid mixed with blood. On the basis of cytological features, the diagnosis of? Hyperplastic goitre with hypothyroidism was rendered (Figures 2a & Figure 2b). However, the possibility of follicular patterned malignancy (Bethesda category-3) was not ruled out for which histopathological examination was advised. Right hemithyroidectomy was done and the patient was put on hormone replacement, post-operatively.
Figure 2a: follicular cells arranged I’m microfollicular pattern in a background of scant colloid.
Figure 2b: Follicular cells showing mild anisonucleosis.
Grossly, a right hemithyroidectomy specimen measuring 6x5x3cm was received (Figure 3) which was partially encapsulated. Cut section showed three solid firm greyish- white nodular areas which measured 2x1cm, 1x1cm and 0.5x0.3cm respectively (Figure 4). Whole tissue was processed in 05 paraffin blocks.
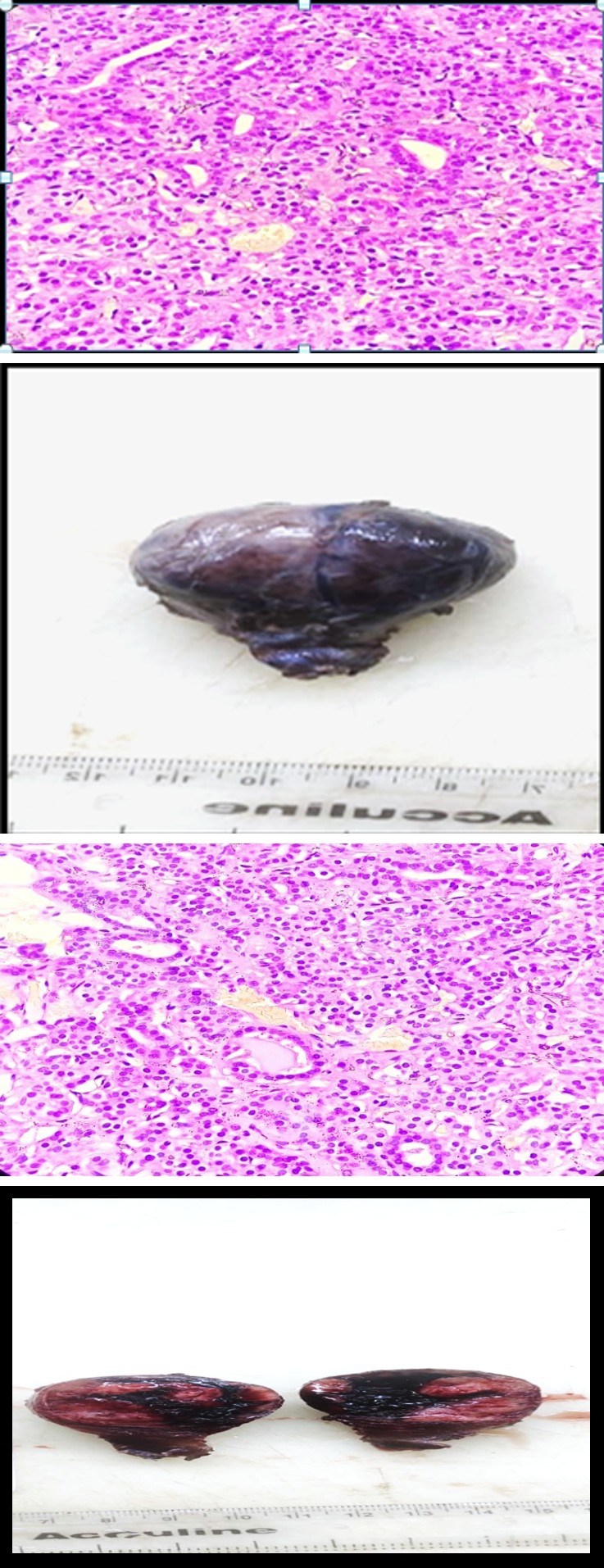
Figure: 3 Gross picture showing hemithyroidectomy specimen.
On histological examination, these nodules were partially encapsulated and showed variable architecture and cellular morphology. Sections showed solid appearance mostly with hyper-cellular nodules having irregular borders comprising of ill-formed follicles with absent to minimal colloid. Some areas showed diffuse sheets of closely packed follicular cells without lumen separated by thick fibrous septa, exhibiting marked cellular pleomorphism with bizarre hyperchromatic nuclei and multi-nucleated giant cells. Inter-nodular thyroid parenchyma also showed bizarre-looking closely packed follicular cells in nests and sheets (Figures 5a & 5b). None of the sections showed normal thyroid parenchyma. The Congo red stain was negative.
Figure 5a: Section shows closely packed follicles with scant colloid (H&E x100).
Figure 5b: Section shows follicular cells with mild nuclear atypia and scant intraluminal colloid (H&E x400).
A differential diagnosis of multi-nodular goitre was ruled out as there was scant to absent colloid in the follicles and absence of distended colloid-filled follicles, instead, there were empty closely packed follicles and sheets of follicular cells exhibiting bizarre pleomorphic follicular cells, mimicking a follicular carcinoma closely. In our case in spite of architectural complexity and cytological atypia, there was no capsular or vascular invasion, hence the diagnosis of follicular carcinoma was ruled out. A final diagnosis of dyshormonogenetic goitre was made excluding the other possible diagnosis, in the context of clinical history and laboratory findings. Intrathyroidal ectopic thymus tissue of 5mm size was found as an incidental finding.
Discussion
Congenital dyshormonogenetic goitre represents about 10-20% of all cases of congenital hypothyroidism. The two most common causes for dyshormonogenesis are: - 1) Defective organification of iodine, frequently the result of mutations in the Thyroid Peroxidase (TPO) gene, and 2) Defective synthesis and secretion of thyroglobulin synthesis. As a result of impaired thyroid hormone synthesis, thyroid stimulating hormone secretion increases, resulting in compensatory goitre. [4] According to recent studies, there is an increase in the prevalence of congenital hyperplasia, predominantly due to dyshormonogenesis, particularly in the Asian population [6]. In a patient of less than 20 years of age, the diagnosis of dyshormonogenetic goitre should be considered before rendering any such lesion the diagnosis of follicular carcinoma [7].
Due to its diversity in radiographic and FNAC morphological findings, there is almost always a diagnostic dilemma in patients with DHG. The cytomorphological features closely resemble follicular lesions and follicular neoplasms. The presence of scant or no colloid with the presence of sometimes atypical cells closely mimics follicular malignancy or even medullary carcinoma [8]. Cytology alone is not able to differentiate dyshormonogenesis from malignancy. Hence, fine needle aspiration alone is not recommended for the diagnosis of dyshormonogenetic goitre. Microscopically DHG is a diffuse process with prominent nodule formation and no or minimal normal appearing thyroid tissue. The nodules are predominantly solid, or micro-follicular, insular, trabecular or microcystic [9]. The inter-nodular thyroid parenchyma is generally hyperplastic with the presence of atypical cells having marked nuclear atypia with bizarre nuclei. There is no or minimum colloid within the entire thyroid lobe. The severity of the microscopic findings also correlates with the severity and duration of gene defects [10]. No vascular or capsular invasion should be present. Thus, any child or adult should be suspected of a diagnosis of Dyshormonogenic Goitre, presenting with the above microscopic findings and clinical features of hypothyroidism.
Treatment of DG required T4 replacement therapy with L-thyroxine since congenital hypothyroidism is revealed. Total or partial thyroidectomy is advised when malignancy is suspected or for cosmetic reasons or when the goitre is too large causing dysphagia or dyspnea [11]. Immunostaining is not needed for diagnosis. DG usually has an excellent prognosis when diagnosed and treated early. Mental retardation and growth restriction are the most common complications [12, 13].
Conclusion
Dyshormonogenetic goitre is a rare inherited disorder affecting the thyroid, being the second most common cause of congenital hypothyroidism after thyroid dysgenesis. The patient presents generally in the first decade of life with clinical signs and symptoms of hypothyroidism and goitre along with growth and mental retardation. Differential diagnosis includes multinodular goitre, graves’ disease, dyshormonogenetic goitre, radiation thyroiditis, iatrogenic goitre, papillary carcinoma and follicular carcinoma. Based on clinical and cytopathological features only a suggestion of follicular thyroid nodular disease favouring multinodular goitre can be given. However, it is only after the histopathological examination a diagnosis of dyshormonogenetic goitre can be made with certainty hence it becomes imperative to get a thorough histopathological examination of the excised thyroid tissue knowing accurate morphological patterns of a dyshormonogenetic goitre to avoid serious diagnostic pitfall of follicular carcinoma.
References
-
Clerc J, Monpeyssen H, Chevalier A, Amegassi F, Rodrigue D, et al. (2008) Scintigraphic imaging of paediatric thyroid dysfunction. Horm Res Paediatr 70: 1-13.
-
Bchir A, Mestiri S, Belakhdhar M, Abdelkefi M, Mokni M (2022) Dyshormonogenetic goiter: A clinico-pathologic study of a rare disease with literature. Int J med sci clin Res 8(4): 24-28.
-
Braham E, Rejeb HB, Marghli A, Kilani T, El Mezni F (2013) A rare and particular form of goiter to recognize. Ann Transl Med 1: 1-4.
-
Park SM, Chatterjee VK (2005) Genetics of congenital hypothyroidism. J Med Genet 42: 379-389.
-
Wasniewska M, Salerno M, Cassio A, Corrias A, Aversa T, et al. (2009) Prospective evaluation of the natural course of idiopathic subclinical hypothyroidism in childhood and adolescence. Eur J Endocrinol 160: 417-421.
-
Yu B, Long W, Yang Y, Wang Y, Jiang L, et al. (2018) Newborn screening and molecular profile of congenital hypothyroidism in a Chinese population. Front Genet 9: 509-510.
-
Schwartz MR (1990) Pathology of the thyroid and parathyroid glands. Otolaryngol. Clin North Am 23: 175- 215.
-
Ghossein RA, Rosai J, Heffess C (1997) Dyshormonogenetic goiter: a clinicopathologic study of 56 cases. Endocr. Pathol 8: 283-292.
-
Baloch ZW, LiVolsi VA (2006) Cytologic and architectural mimics of papillary thyroid carcinoma: diagnostic challenges in fine-needle aspiration and surgical pathology specimens. Pathol. Patterns Rev 125: 135-144.
-
Medeiros-Neto G, Stanbury JB (2019) Inherited disorders of the thyroid system. CRC Press control ser 1-232.
-
Fisher DA, Klein AH (1981) Thyroid development and disorders of thyroid function in the newborn. N Engl J Med 304: 702-712.
-
Matos PS, Bisi H, Medeiros-Neto G (1994) Dyshormonogenetic goiter: A morphological and immunohistochemical study. Endocr Pathol 5: 49-58.
-
Yashiro T, Ito K, Akiba M, Kanaji Y, Obara T, et al. (1987) Papillary carcinoma of the thyroid arising from dyshormonogenetic goitre. Endocrinol Japon 34: 955- 964.
- Genomic Landscape of Aggressive Penile Squamous Cell Carcinoma including TERT-p and NOTCH1 Mutations – An Institutional Experience
- Establishment of Baseline Haematological Values for Canine Population in North-Central Nigeria: A Cross-Sectional Study in the Federal Capital Territory
- Biochemical Assessment of Uroliths Extracted in Patients with Urolithiasis in a Tertiary Health Institution
- Update on Gastrointestinal Pecomas: Molecular Pathogenesis and Risk Stratification
- A Comparative Study of Serum C-reactive Protein Level Between Pre-eclampsia and Normal Pregnancy in Tertiary Level Hospital
- From Deformity to Alignment: Clinical Outcomes of the Schnepp Osteotomy in Hallux Valgus in 47 Feet