Differential Correlation between Foetal Haemoglobin and Full Blood Count Based on Inherited Haemoglobin Type; A Cross-Sectional Study in Cape Coast, Ghana
Background: Foetal haemoglobin (Hb F) has been shown to modulate the severity of sickle cell anaemia (SCA). However, there is scarcity of data on the impact of Hb F levels in other inherited haemoglobin variants. This cross-sectional study sought to investigate the relationship between Hb F and full blood count parameters in participants based on their inherited haemoglobin type. Materials and methods: Four milliliters of venous blood were drawn from 170 consecutively consented participants (aged 10 -55 years) into EDTA-anticoagulated tubes. Full blood counts (FBC) were estimated using Horiba ABX Pentra XL80 analyzer, whereas haemoglobin variants were determined using cellulose acetate electrophoresis. Hb F was estimated using the modified Bekte-alkali denaturation method. Data was analysed using SPSS (version 25 for Windows). Relationship between Hb F and FBC parameters were explored using Pearson correlation coefficient analyses. Statistical significance was established at p Results: Whereas majority (61.8%) were females, there was no significant differences in age among the participants based on gender. Participants with inherited haemoglobin variants comprised 20% of the study population. Total WBC was significantly higher in participants with inherited haemoglobin variants (p = 0.011). Hb F levels were also significantly elevated in participants with inherited haemoglobin variants (p
Introduction
Globally, haemoglobinopathies are one of the most common genetic inherited disorders. Previous study by Kohn estimated prevalence of 5 – 30% in Africans, 5 – 40% in Arabians, 5 – 20% Central Americans, and 0.5 – 1% in Europeans [1]. Although, it has been stated that selective pressures due to severe malaria infection may partly account for the high prevalence of haemoglobinopathies in sub-Saharan Africa, it is still important to mention that effective premarital genetic counselling can significantly reduce the disease burden [2]. Sickle cell anaemia (SCA) is perhaps the most studied of the haemoglobin variants probably because of the severe vaso-occlusive crises that it causes to those with homozygous Hb SS inheritance [3]. The other haemoglobin variants like Hb C, D, E etc. are not as well studied even though the inheritance of any of these may impact varying rheological properties to the red cells. Foetal haemoglobin (Hb F) has been demonstrated both experimentally and clinically to modulate severity of SCA [4, 5, 6]. The relationship between the various haemoglobin variants and some blood components have been established by other studies. For instance, the prevalence and influence of HbS, HbC and Hb- Thalassemia on red blood cell parameters has been reported previously [7]. The interaction of the various Hb variants with white blood cells remains unclear [8]. The role Hb F levels play in modulating the FBC parameters in other haemoglobin variants have also not been clearly delineated. We thus sought to interrogate the relationship between the Hb F levels and full blood count parameters in participants with inherited haemoglobin variants as well as those without any inherited haemoglobin variants. The aim was to investigate whether the relationship established for individuals with SCA extends to the other haemoglobin variants considering that the inheritance of any of these different haemoglobin variants may impact different rheological properties to the red blood cells.
Materials and Methods
Study Design/Population
This cross-sectional study consecutively recruited 170 consenting participants aged 10 – 55 years. The participants attended the out-patient department (OPD) section of the University of Cape Coast Hospital, Ghana, between January 2017 – May 2017.
Data Collection
Four (4) milliliters of venous blood was drawn from each participant into EDTA-anticoagulated tubes following standard protocols. The blood was used for haemoglobin electrophoresis, %Hb F and and full blood count (FBC).
Ethical Consideration
All protocols for the study were approved by University of Cape Coast Institutional Review Board (IRB number, UCCIRB/CHAS/2017/76), Department of Medical Laboratory Technology and the University of Cape Coast hospital.
Full Blood Count (FBC)
FBC for each sample was estimated using Horiba ABX Pentra XL80 (Horiba medical, Japan) automated haematology analyser. The RBC count, red cell indices, haemoglobin (Hb) concentration, Platelet count and WBC count and WBC absolute differential counts were recorded.
Estimation of Fetal Hemoglobin (Hb F)
Concentration of Hb F was estimated for each sample according to previously described protocols Bekte, et al. Samples were centrifuged at 2000 rpm for 10 minutes to remove plasma before haemolysates were prepared. The proportion of Hb F was calculated from the absorbance ratio between Hb F and total Hb after correcting for the dilution factor as: Hb F (%) = [100*Absorbance of test/ (Absorbance of Ref*20)].
Cellulose Acetate Haemoglobin Electrophoresis
Hb variant of each participant was determined by electrophoresis on cellulose acetate using whole blood in accordance with previously published protocols [9]. A control sample containing haemoglobin (Hb) A, C, S, and F was run with each sample to ensure validity of results interpretation.
Data Analysis
Data collected was entered into Microsoft Office Excel 2016 and analysed using Statistical Package for Social Sciences (SPSS) version 25.0 for Windows (IBM, USA). A summary was presented using descriptive statistics such as frequencies, percentages, mean and standard deviations. Chi square analysis was used to establish association between Hb variants and gender. Pearson correlation coefficient analysis was used to explore the relationship between %Hb F levels and FBC parameters. One-Way ANOVA (with Dunnett’s multiple comparison) was used to compare the absolute white blood cell differential counts between Hb AA participants and those with inherited haemoglobin variants. A p value of <0.05 was considered statistically significant.
Results
Table 1 describes the general characteristics of the study participants. Whereas majority belonged to the 21 – 30 years age group, 61.2% were females. There was no significant difference in participant age based on gender. Also, 20% inherited some form of hemoglobin variant. Leukocytosis was observed in 10.0% of the participants with 2.9% demonstrating thrombocytosis. Additionally, 17.1% of the participants had %Hb F ≥2.5 (7.7% males vs 22.9% females).
| Parameter | Total | Males | Females | p-value |
| Parameter | N(%) | N=65 | N=105 | p-value |
| Age(years) | 0.971 | |||
| 10-20 | 45(26.5) | 17(26.2) | 28(26.7) | |
| 21-30 | 59(34.7) | 22(37.3) | 37(62.7) | |
| 31-40 | 30(17.6) | 11(16.9) | 19(18.1) | |
| >40 | 36(21.2) | 15(23.1) | 21(20.0) | |
| Hb variants | ||||
| A | 136(80.0) | 52(80.0) | 84(80.0) | 0.761 |
| AS | 18(10.6) | 6(9.2) | 12(11.4) | |
| AF | 5(2.9) | 3(4.6) | 2(1.9) | |
| SS | 2(1.2) | 0(0.0) | 2(1.9) | |
| AC | 7(4.1) | 3(4.6) | 4(3.8) | |
| SC/CC | 2(1.2) | 1(1.5) | 1(1.0) | |
| WBC(*10^9/L) | 0.148 | |||
| <3.0 | 5(2.9) | 4(6.2) | 1(1.0) | |
| 3.0-11.0 | 148(87.1) | 55(86.2) | 93(88.6) | |
| >11.0 | 17(10.0) | 1(1.5) | 11(10.5) | |
| Platelet(*10^9/L) | 0.083 | |||
| <150 | 12(7.1) | 8(12.3) | 4(3.8) | |
| 150-450 | 153(90) | 56(86.2) | 97(92.4) | |
| >450 | 5(2.9) | 1(1.5) | 4(3.8) | |
| %Hb F | 0.012 | |||
| <2.5 | 141(82.9) | 60(92.3) | 81(77.1) | |
| ≥2.5 | 29(17.1) | 5(7.7) | 24(22.9) |
Table 1: General characteristics of study participants.
Table 2 compares the mean FBC parameters in relation to the type of inherited hemoglobin variant. The RBC count did not significantly differ across the different inherited haemoglobin types. The WBC count was significantly higher in participants with Hb AS, AF, SS, or AC compared to those with haemoglobin A (p = 0.011). Also, the %Hb F significantly differed across participants with different haemoglobin types. However, platelet count did not significantly differ across the different haemoglobin types.
| HB Variants | Mean (±Std) | P-Value | |
|---|---|---|---|
| RBC | 0.242 | ||
| AA (136) | 4.529(±0.666) | ||
| AS (18) | 4.488(±0.784) | ||
| AF (5) | 4.666(±0.561) | ||
| SS (2) | 4.825(±0.926) | ||
| AC (7) | 4.980(±0.746) | ||
| SC/CC (2) | 5.430(±0.566) | ||
| WBC | 0.011 | ||
| AA (136) | 6.542(±2.633) | ||
| AS (18) | 10.395(±11.465) | ||
| AF (5) | 10.060(±4.111) | ||
| SS (2) | 11.350(±1.061) | ||
| AC (7) | 11.186(±16.137) | ||
| SC/CC (2) | 3.950(±1.768) | ||
| HbF | <0.0001 | ||
| AA (136) | 0.728(±0.465) | ||
| AS (18) | 1.206(±0.768) | ||
| AF (5) | 16.044(±11.412) | ||
| SS (2) | 1.625(±0.997) | ||
| AC (7) | 0.940(±0.927) | ||
| SC/CC (2) | 2.150(±0.636) | ||
| PLT | 0.567 | ||
| AA (136) | 268.934(±90.885) | ||
| AS (18) | 274.167(±67.116) | ||
| AF (5) | 274.400(±74.614) | ||
| SS (2) | 172.500(±132.229) | ||
| AC (7) | 262.000(±49.608) | ||
| SC/CC (2) | 195.000(±74.953) |
Table 2: Comparison of mean HB, WBC, HBF and PLTs with the hemoglobin variants of participants.
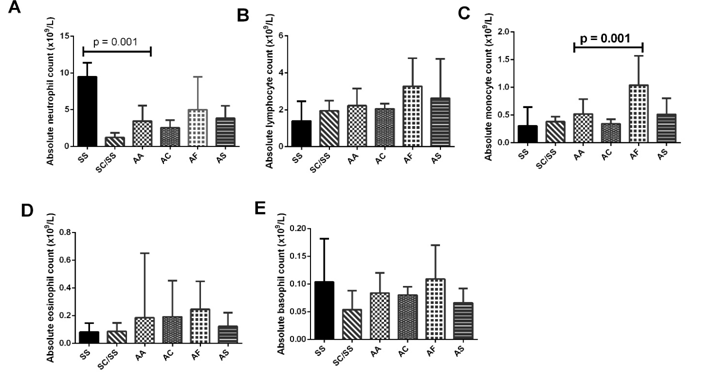
Participants differential WBC were compared based on their inherited haemoglobin type (Figure 1). Although neutrophil counts differed across the haemoglobin types, only participants with Hb SS had significantly higher neutrophil count compared to Hb AA participants. Also, monocyte counts were significantly higher in Hb AF participants compared to those with Hb A. Although basophil, lymphocyte, and lymphocyte counts differed across the haemoglobin types, these did not reach statistical significance. This study also investigated the relationship between Hb F levels and red cell indices (Tables 3). Whereas Hb F was inversely related to all red cell indices (reaching significance in Hb and HCT) in participants with no haemoglobin variants, it was directly related to all red cell indices (reaching significance in Hb, HCT, and MCH) in participants with inherited haemoglobin variants. Additionally, whereas Hb F was directly related to platelet counts in participants with Hb A, it was inversely related with platelet count in participant with inherited haemoglobin variants.
| Parameter | Hb A | Hb variants | ||||
|---|---|---|---|---|---|---|
| AA (136) | 6.542(±2.633) | |||||
| AS (18) | 10.395(±11.465) | |||||
| AF (5) | 10.060(±4.111) | |||||
| SS (2) | 11.350(±1.061) | |||||
| AC (7) | 11.186(±16.137) | |||||
| SC/CC (2) | 3.950(±1.768) | |||||
| HbF | <0.0001 | |||||
| AA (136) | 0.728(±0.465) | |||||
| AS (18) | 1.206(±0.768) | |||||
| AF (5) | 16.044(±11.412) |
Table 3: Pearson correlation between Hb F and red cell indices among participants.
| Parameter | Hb A | Hb variants | ||||
|---|---|---|---|---|---|---|
| SC/CC (2) | 2.150(±0.636) | |||||
| PLT | 0.567 | |||||
| AA (136) | 268.934(±90.885) | |||||
| AS (18) | 274.167(±67.116) | |||||
| AF (5) | 274.400(±74.614) | |||||
| SS (2) | 172.500(±132.229) | |||||
| AC (7) | 262.000(±49.608) | |||||
| SC/CC (2) | 195.000(±74.953) | |||||
Table 4: Pearson correlation between Hb F and WBC differentials in study participants.
Discussion
In sub-Saharan Africa, selective pressure due to malaria has led to balanced polymorphism that selects for some genetically inherited traits that gives survival advantages. Thus, evolution has preserved genetically inherited diseases such as sickle cell disease in sub- Saharan Africa. Research has demonstrated that increased Hb F expression has co-evolved with SCD to mitigate the severity of this disease. In this study, we showed that 20% of the participants inherited some form of variant haemoglobin, with 17.1% demonstrating elevated %Hb F levels. Additionally, whereas those with inherited haemoglobin variants demonstrated inverse correlation between % Hb F and WBC or platelet count, those with no inherited variant haemoglobin generally showed direct correlation between Hb F and WBC or platelet count. Within the limits of the cellulose acetate electrophoresis technique, we report that a wide spectrum of variant haemoglobin (Hb S, C, F) were detected in the study population. The limitations associated with cellulose acetate electrophoresis is well documented in the literature particularly the co- migration of some haemoglobin variants [10, 11]. Therefore, it is possible that this study underestimated the spectrum of variant haemoglobin in the study population. The prevalence of haemoglobin variant inheritance in sub-Saharan Africa has been stated at 5% - 30% [1]. In Ghana specifically, previous cross-sectional studies have found 25 – 30% prevalence of haemoglobin variant inheritance [12, 13]. This cross-sectional study which used convenience sampling design found a prevalence of 20% among the study population which is comparable to the estimated prevalence for the sub- region. Considering that premarital genetic counselling could significantly reduce the incidence of haemoglobinopathies [2], this high prevalence is indicative that premarital counselling is probably not being given adequate consideration in our study population. Sickle cell trait was the predominant haemoglobin variant detected in the study population. Elevated Hb F levels in individuals with SCA have been extensively reported in the literature [5, 14, 15]. This study demonstrated that increased Hb F production is not limited to SCA, but also other variant haemoglobin as well. This study employed the Bekte-alkali denaturation method to report elevated Hb F levels comparable to that of other reports in the sub-region that used the same technique in similar study designs [14, 16]. Total WBC counts were significantly elevated in participants with inherited haemoglobin variants compared to those without. This is in agreement with previous findings by Akinlosotu, et al. that reported significantly elevated WBC in sickle cell anaemia children in South-Western Nigeria [17]. However, whereas Akinlosotu also reported significant differences in the differential WBC parameters, we could only establish significant increases in monocytes and neutrophil counts in Hb AF and AS respectively. The slightly different study designs might have accounted for this. Whereas the study by Akinsolotu compared the differential WBC between Hb AA individuals with that of individuals inheriting only Hb SS in children (50% cases against 50% controls), this study largely recruited adult population and with those inheriting haemoglobin variants comprising only 20% of the participants. Individual with SCA comprised only 2.4% of the study population in the present study (compared to 50% in the Akinlosotu study). It is interesting to point out that leukocytosis reported herein has also been demonstrated in several multi-center studies as a risk-factor for severe sickle cell disease- related complications including death [18, 19, 20]. This poor prognostication of leukocytosis is supported in the present study by the inverse relationship between Hb F and leucocyte count established among participants with inherited haemoglobin variants. This study also found significant differential Hb F correlations with red cell indices or platelet count between those with inherited haemoglobin variants and those without. The direct associations between red cell indices and Hb F recorded among the participants with inherited Hb variants has been previously reported in another cross-sectional study in the sub-region [17]. Indeed, it has been demonstrated by others that elevated Hb F levels are protective in SCA patients [3, 6, 21, 22]. The fact that high leucocyte count has been identified in multivariate analyses as powerful predictor of favourable response to hydroxyurea therapy may be an indication that the driving force behind this evolved mechanism may not be red cell indices. This is buttressed by the direct correlation between Hb F and red cell indices in those with inherited Hb variants contrary to the observation in Hb AA clients. On the other hand, since those with no haemoglobin variants are not under the same selective pressures as those with inherited haemoglobin variants, different mechanisms might be at play to regulate Hb F expression. The inverse association between Hb F and platelet count, although reported in another study [17], requires further studies to clearly delineate the role platelets play in the pathogenesis of haemoglobinopathies.
Conclusion
Leukocytosis and inverse relationship between Hb F and WBC or platelet count in those with haemoglobin variants might predispose these individuals to severe manifestations of haemoglobinopathy. Acknowledgement: We are very grateful to the staff of the medical laboratory unit of the University of Cape Coast Hospital for their support throughout the period of the research. We are also indebted to the patients who consented to be part of the study. Author contributions: PA conceived, designed, supervised all the experimental work in the research and revised the manuscript. FA, DB, and RA were involved in participant recruitment, sampling, and experimental work. BKSD analysed the data and wrote the initial draft. RPS was involved in literature search, initial manuscript editing and experimental work. All authors read and approved the final manuscript prior to submission. Conflict of interest: The authors declare no conflict of interest. Funding: The authors received funding from no agency and funded the research themselves.
References
-
Kohne E (2011) Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int 108(31-32): 532-540.
-
McGann PT, Hernandez AG, Ware RE (2017) Sickle cell anemia in sub-Saharan Africa: advancing the clinical paradigm through partnerships and research. Blood 129(2): 155-161.
-
Antwi-Boasiako C, Frimpong E, Ababio GK, Dzudzor B, Ekem I, et al. (2015) Sickle Cell Disease: Reappraisal of the Role of Foetal Haemoglobin Levels in the Frequency of Vaso-Occlusive Crisis. Ghana Med J 49(2): 102-106.
-
Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, et al. (1992) Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 79(10): 2555-2565.
-
Dover GJ, Smith KD, Chang YC, Purvis S, Mays A, et al. (1992) Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood 80(3): 816- 824.
-
Goldberg MA, Brugnara C, Dover GJ, Schapira L, Lacroix L, et al. (1992) Hydroxyurea and erythropoietin therapy in sickle cell anemia. Semin Oncol 19(3): 74-81.
-
Kohne E (2011) Clinical Manifestations, Diagnosis, and Treatment significance 5(11): 12.
-
Olatunji BO, Davis ML, Powers MB, Smits JA (2013) Cognitive-behavioral therapy for obsessive- compulsive disorder: a meta-analysis of treatment outcome and moderators. J Psychiatr Res 47(1): 33- 41.
-
Cheesbrough M (2006) District laboratory practice in tropical countries. Cambridge university press.
-
Assendelft OW (2002) Detection and identification of haemoglobin variants, in Advanced Laboratory Methods in Haematology, Rowan PFE, Martin R, (Eds.), Arnold Publishers: Oxford, UK, pp: 195-206.
-
Adu P, Nicodemus LS, Kate Kontor, Richrd KD Ephraim (2017) Misleading presentation of haemoglobin electrophoresis data. Ghana Med J 51(1): 36-38.
-
Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, et al. (2011) Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med 41(6): 398-405.
-
Ohene-Frempong K, Joseph O, Hannah T, Francis N (2008) Screening newborns for sickle cell disease in Ghana. Pediatrics 121(2): 120-121.
-
Omoti CE (2005) The value of foetal haemoglobin level in the management of nigerian sickle cell anaemia patients. Niger Postgrad Med J 12(3): 149- 154.
-
Economou EP, Antonarakis SE, Kazazian HH, Serjeant GR, Dover GJ (1991) Variation in hemoglobin F production among normal and sickle cell adults is not related to nucleotide substitutions in the gamma promoter regions. Blood 77(1): 174-177.
-
Olaniyi JA, Arinola OG, Odetunde AB (2010) Foetal haemoglobin (Hb F) status in adult sickle cell anaemia patients in Ibadan, Nigeria. Ann Ib Postgrad Med 8(1): 30-33.
-
Akinlosotu MA, Adegoke SA, Oseni SB, Adeodu OO (2017) Relationship between foetal haemoglobin and haematological indices in children with sickle cell anaemia from South Western Nigeria. Niger Postgrad Med J 24(4): 195-200.
-
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, et al. (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 330(23): 1639-1644.
-
Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, et al. (1998) Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91(1): 288-294.
-
Powars DR (2000) Management of cerebral vasculopathy in children with sickle cell anaemia. Br J Haematol 108(4): 666-678.
-
Dover GJ, Charache S (1992) Hydroxyurea induction of fetal hemoglobin synthesis in sickle-cell disease. Semin Oncol 19(3): 61-66.
-
McDonagh KT, Dover GJ, Donahue RE, Nathan DG, Agricola B, et al. (1992) Hydroxyurea-induced HbF production in anemic primates: augmentation by erythropoietin, hematopoietic growth factors, and sodium butyrate. Exp Hematol 20(10): 1156-1164.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies