Expanding the Classification of Leukemia by the World Health Organization over Time
The classification has always been the basis for many treatments of the disease. The French-American-British has performed leukemia classification base on morphological finding, but the World Health Organization apply gene or chromosomal mutations in its classification. This approach has revolutionized the diagnosis, choose strategy of treatment and follow-up ever since. Although WHO has classified leukemia on the basis of FAB; but genetic mutations has made the wider and more specific outcome. Obviously, identifying these damages provide an extensive filed for researchers and physicians to effective assistance for patients. Therefore, the purpose of this review is comparing revisions of WHO classification in the years 2008 and 2016, because these categories are highlighted unknown cases that give researchers incentives to further study.
Introduction
The purpose of each classification of hematological neoplasms is to provide reliable criteria for diagnosis and also appropriate classification of them. These classifications has changed over time. Surprisingly, given the advancement of molecular study and the identification of newer and more specific factors, changes to these categories occur simultaneously with changes in advances science. In 1982, French-American-British (FAB) group presented a classification by using morphological and cytochemical characteristic of blast cells. By developing diagnostic methods and identifying molecular aspects of leukemia, the World Health Organization (WHO) expanded this classification by maintaining the FAB classification. The basic principle of the WHO is that information about, genetic, immunophenotypic, biologic, and clinical features along with morphology should be considered, in order to accurately diagnose and determine the protocol of treatment based on it. Also in WHO classification the number of blasts necessary for the diagnosis is more than 20% in bone marrow when compared 30% in FAB classification. Since 2001, when the WHO presented this report, these categories have been updated and updated. The latest changes are for 2016. Based on these concepts, the progression of the diagnostic criteria, it is anticipated that prognosis, quality of life and patient survival will improve in the future. This review attempts to compare these changes over the years. Also focus on known mutation during this time and emphasis on the search for unknown mutations.
Classification of MPNs have not changed remarkably since the 2008 edition, but discoveries of new mutations and improved understanding of the morphologic characteristics of some entities have impacted the diagnostic criteria for the disease entities. Table 1 lists diseases encompassed in the MPNs and compares the 2016 revision with that of the 2008 classification.
Myeloproliferative Neoplasm (MPN)
| 2008 | 2016 | ||||
|---|---|---|---|---|---|
| C | hronic myelogenous leukemia(CML), BCR-ABL1 | + | Chronic myeloid leukemia(CML), BCR-ABL1+ | ||
| Chronic Neutrophilic Leukemia (CNL) | Chronic Neutrophilic Leukemia (CNL) | ||||
| Polycythemia Vera (PV) | Polycythemia Vera (PV) | ||||
| Primary Myelofibrosis (PMF) | Primary Myelofibrosis (PMF) | ||||
| PMF, prefibrotic/early stage | |||||
| PMF, overt fibrotic stage | |||||
| Essential Thrombocythemia(ET) | Essential Thrombocythemia (ET) | ||||
| Chronic Eosinophilic Leukemia, NOS | Chronic Eosinophilic Leukemia not otherwise specified, (CEL-NOS) | ||||
| Mastocytosis Myeloproliferative Neoplasm | |||||
| Mastocytosis unclassifiable (MPN-U) | |||||
| unclassifiable | |||||
Table1: The 2008 and 2016 WHO Classification of MPNs.
Chronic Myeloid Leukemia (CML)
Based on the 2016 update of WHO classification the "myelogenous" is replaced with "myeloid" in CML. So the name of chronic myelogenous leukemia is changed to myeloid leukemia.
By identifying the BCR-ABL1 fusion gene in this group of patients, tyrosine kinase inhibitor is considered as a first-line treatment. In the new classification criteria for diagnosis, accelerated-phase (AP) was revised. So “response to tyrosine kinase inhibitor (TKI)” criteria have been added to the definition of AP disease. These include: a) Hematologic resistance to first TKI b) Hematologic, cytogenetic, or molecular indications of resistance to two sequential TKIs c) Occurrence of two or more mutations in BCR-ABL1 during TKI therapy [1]. Table 2 shows Response criteria of TKI therapy [2]
| Time on TKI therap | y | ELN /NCCN recommendations | |||||
|---|---|---|---|---|---|---|---|
| Optimal | Warning | Failure | |||||
| baseline | NA | High risk or CCA/Ph +, major route | NA | ||||
| 3 months | BCR-ABL1 ≤ 10% and/or Ph+ ≤ 35% | BCR-ABL1 > 10% and/or Ph+ 36- 95% | Non-CHR and/or Ph+ >95% | ||||
| 6 months | BCR-ABL1 ≤ 1% and/or Ph+ 0 | BCR-ABL1 1- 10% and/or Ph+ 1- 35% | BCR-ABL1 > 10% and/or Ph+ >35% | ||||
| 12 months | BCR-ABL1 ≤ 0.1% | BCR-ABL1>0.1-1% | BCR-ABL1 >1% and/or Ph+ >0 | ||||
| 18 months | |||||||
| Then, and at any time | BCR-ABL1 ≤0.1% | CCA / Ph-(-7, or 7q-) | Loss of CHR, Loss of CCgR, Confirmed loss of MMR Mutations CCA/ Ph+ |
Table 2: Response criteria of TKI therapy.
ELN: European Leukemia Net; NCCN: National Comprehensive Cancer Network; NA: not available; CCA: Clonal Chromosome Abnormalities; CHR: Complete Hematological Response defined as WBC < 10 × 109/L, Basophils <5%, No myelocytes, promyelocytes, myeloblasts in the differential, Platelet count <450 ×109/L, Spleen non-palpable; CCgR: Complete Cytogenetic Response defined as no Ph+ metaphases on chromosome banding analysis (at least 20 bone marrow cell metaphases) or <1% BCR-ABL1 positive nuclei of at least 200 nuclei on fluorescence in situ hybridization. MMR: Major Molecular Response defined as BCR-ABL1 expression of ≤0.1% on the international scale. Table 2: Response criteria of TKI therapy.
Chronic Neutrophilic Leukemia (CNL)
CNL is a clonal disorder in which neutrophils proliferate uncontrollably. In the new category in 2016, diagnostic cases of CNL are divided into the major and minor criteria. Also, in the new classification, presence of colony-stimulating factor 3 receptor (CSF3R) T618I or other membrane-proximal CSF3R mutations have been added to CNL criteria diagnosis [3, 4]. CSF3R is the primary growth factor of neutrophil production. The CSF3R mutation has two categories: truncation mutations and membrane proximal mutations. CSF3R membrane proximal mutations may be sensitive to JAK kinase inhibitors such as ruxolitinib. While truncation mutations displayed sensitivity to inhibition with SRC kinase inhibitor Dasatinib [4, 5].
Polycythemia Vera (PV)
Polycythemia Vera is one of the Philadelphia chromosome-negative myeloproliferative neoplasm which is characterized by proliferation of erythroid, granulocytic and megakaryocytic elements in the bone marrow that are called panmyelosis. According to the 2016 WHO classification, due to the importance of red cell mass (RCM) parameter and the effect of hematocrit on it, the Hct parameter was considered as a suitable measure for the detection of PV. The optimal cutoff of hematocrit was considered 49% in men and 48% in women, and the hemoglobin level of 16.5g / dl and 16g/ dl in men and women respectively has been determined for distinguishing masked/prodromal PV patients from JAK2 mutated ET. According to the new revision the level of EPO as the only minor criterion is considered. Whereas a low serum EPO is specific for PV and therefore clinically useful, approximately 20% of PV patients present with normal and a few even with high EPO values [6, 7, 8]. Based on the 2016 WHO classification, bone marrow morphology has been considered as a major criterion for detecting PV. A slight increase in reticulin levels has been observed in patients with bone marrow biopsy. Increased reticulin fibrosis is associated with the progression of the disease to Post PV myelofibrosis [8, 9]. The endogenous erythroid colony (EEC) in the 2016 classification has been eliminated due to time and cost [9].
Essential Thrombocythemia (ET)
ET is a clonal stem cell disease and one of the Philadelphia chromosome-negative myeloproliferative neoplasms which is associated with persistent thrombocytosis. According to the classification of 2016, MPL (the thrombopoietin receptor) and CALR (Calreticulin) mutations have been added to ET diagnostic criterion. 10-16% of ET cases are triple negative. Compared to JAK2-mutated cases, the association of CALR mutation with younger age, male gender, lower leukocyte count, lower Hb level and higher platelet counts is seen, and the thrombotic risk is also lower in them. In the 2016 classification increased levels of reticulin fibers (grade 1) have been observed in major criteria [10].
Primary Myelofibrosis (PMF)
PMF is a Philadelphia-negative myeloproliferative neoplasm characterized by stem cell-derived clonal myeloproliferation with a predominant proliferation of Megakaryocytes and granulocytes in the bone marrow. Bone marrow is filled with fibrous tissue, although in some cases the bone marrow is hyperplastic and very little fibrosis is seen [11, 12]. PMF is categorized into Pre- PMF and Overt-PMF (based on the 2016 WHO classification) which may have a major effect on accurate diagnosis and outcome and prognosis. This distinction is important because prePMF can be confused with ET because of similar symptoms. Detection based on poor prognosis, increased mortality and leukemic transformation rate for prePMF compared with ET. In the 2016 classification CALR and MPL mutation were added to major criteria of PMF [1, 12]. Table 3 compares 2008 and 2016 WHO criteria for PMF.
| 2008 | 2016 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-PMF | Overt-PMF | ||||||||
| Major Criteria | Megakaryocyte proliferation and atypia accompanied by either reticulin and/or collagen fibrosis; or in absence of reticulin fibrosis, megakaryocyte changes must be accompanied by increased marrow cellularity, granulocytic proliferation, and often decreased erythropoiesis (i.e., pre-fibrotic | Megakaryocytic proliferation and atypia, without reticulin fibrosis grade 1, accompanied by increased age-adjusted bone marrow cellularity, granulocytic proliferation and often decreased erythropoiesis | Megakaryocyte proliferation and atypia accompanied by either reticulin and/or collagen fibrosis (grade 2 or 3) |
| PMF) | |||
|---|---|---|---|
| Not meeting WHO criteria for CML, PV, MDS, or other myeloid neoplasm | Not meeting WHO criteria for BCR-ABL1+ CML, PV, ET, MDS or other myeloid neoplasm. | Not meeting WHO criteria for BCR-ABL1+ CML, PV, ET, MDS or other myeloid Neoplasm | |
| Demonstration of JAK2V617F or other clonal marker; in absence of clonal markers, no evidence of secondary bone marrow fibrosis | Presence of JAK2, CALR or MPL mutation or in the absence of these mutations, presence of another clonal marker or absence of minor reactive bone marrow reticulin fibrosis | Presence of JAK2, CALR or MPL mutation or in the absence of these mutations, presence of another clonal marker or absence of minor reactive bone marrow reticulin fibrosis | |
| Minor criteria | Leukoerythroblastosis | Presence of one or more of the following: Anemia not attributed to a comorbid condition; | Presence of one or more of the following: Anemia not attributed to a comorbid condition |
| Increased serum lactate dehydrogenase Anemia | Palpable splenomegaly, | Palpable splenomegaly | |
| Palpable splenomegaly | Leukocytosis ≥11 × 109/L Elevated LDH | Leukocytosis ≥11 × 109/L Elevated LDH Leukoerythroblastosis |
Table 4: Comparison of 2008 and 2016 criteria of PMF.
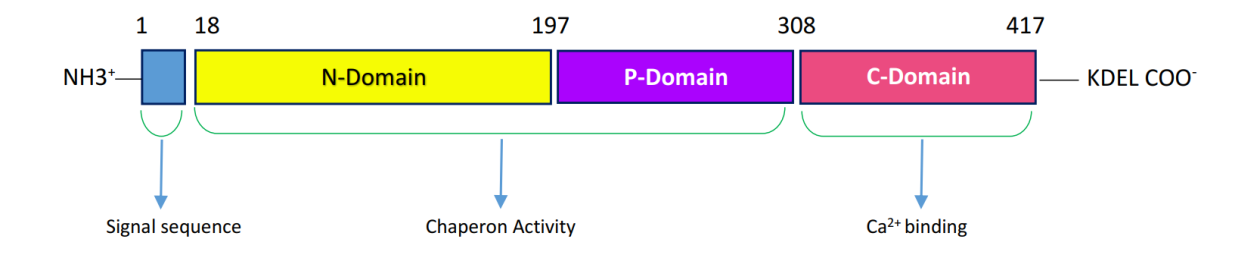
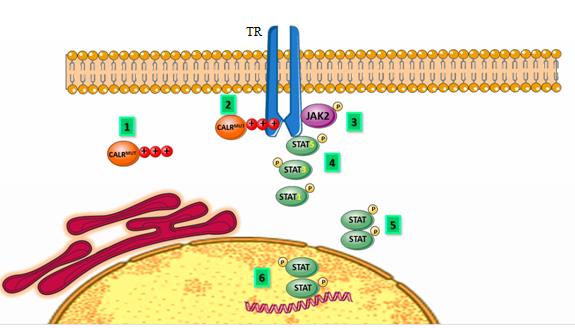
The common mutation types are type 1 mutation (p, L367fs*47) which results from 52-bp deletion, and type 2 mutation (p.K385fs*47) which results from a 5-bp TTGTC insertion within exon 9 of the gene. Both types of mutations cause a single base pair frame shift which results in the formation of a new mutant C-Terminal peptide composed of minimal 36 amino acids which replace the 27 amino acids that are lost from the normal sequence. The mutated C-domain contains positively charged amino acids because of eliminating the negative charge; so the Ca2+ binding function is impaired. Type 1 mutations remove all of the negatively charged amino acids but in type 2 mutations almost up to half of the positively charged amino acids are sustained. Also, in both types of mutation the KDEL motif is lost so mutant CALR have an abnormal Ca2+ binding and cell growth [13, 14] (Figures 1 & 2).
Mastocytosis
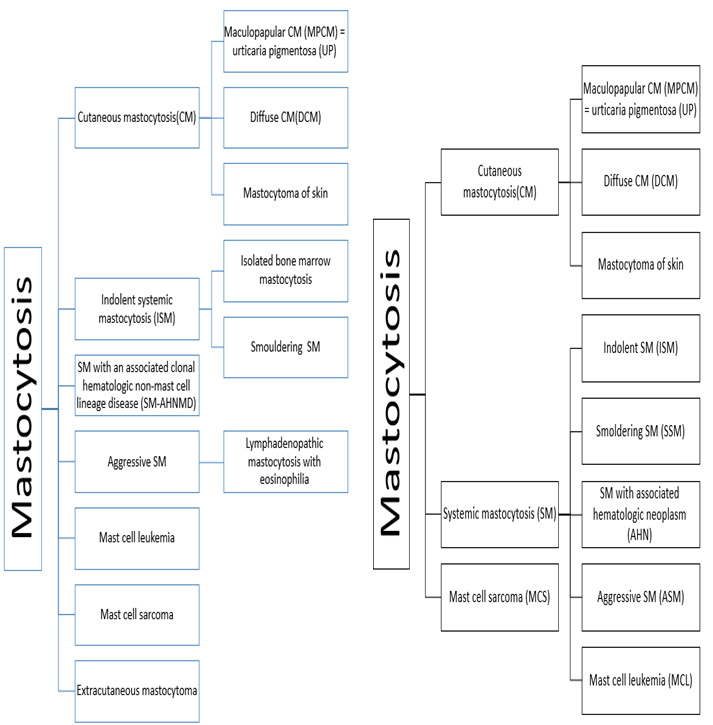
Mastocytosis is a term used for clonal accumulation of mast cells (MCs) in different organs. Most patients with mastocytosis have characteristic skin lesions. Increased levels of mast cell tryptase are considered as an indicator of disease. The WHO update of 2016 recommends use of the following new synonym: AHN (associated hematologic neoplasm) and SM-AHNMD (SM with clonal hematologic non-mast cell lineage disease) and may be used interchangeably. So AHN and SM-AHNMD, can be used interchangeably [15, 16]. In ASM patients, the percentage of Mast cells in bone marrow is important. For this reason, in the new revision of WHO, the disease is divided into two categories: transformed ASM (ASM-t) and untransformed variant. In ASM-t the percentage of MCs in the bone marrow smear is 5-19% and when the percentage of MCs in bone marrow smear increases more than 20%, the diagnosis changes to MCL. In the new revision of WHO classification extra cutaneous mastocytoma has been removed. Smoldering systemic Mastocytosis (SSM) in the new classification is a variant of SM while in the previous category it was a subvariant of indolent SM (ISM) [17] (Figure 3).
Chronic Eosinophilic Leukemia, Not Otherwise Specified (CEL-NOS)
CEL-NOS is a clonal eosinophilia and is considered in the presence of ≥1.5 × 109/L absolute eosinophil count in the peripheral blood more than 6 months. The presence of myeloblast excess (either >2% in the peripheral blood or 5–19% in the bone marrow) and cytogenetic abnormalities such as trisomy 8, t (10; 11) (p14; q21), and t (7; 12) (q11; p11). Recent studies have suggested the possibility of re-classifying some cases of “hypereosinophilic syndrome” as CEL-NOS [18].
Myeloproliferative/Myelodysplastic neoplasm (MPN/MDS)
Myelodysplastic/myeloproliferative neoplasms encompass rare clonal myeloid proliferations that show overlapping myeloproliferative and myelodysplastic characteristics, include chronic myelomonocytic leukemia, atypical chronic myeloid leukemia (BCR-ABL1 negative), juvenile myelomonocytic leukemia (JMML), and myelodysplastic/myeloproliferative neoplasms, unclassifiable [18, 19].
Chronic Myelomonocytic Leukemia (CMML)
Chronic myelomonocytic leukemia (CMML) combines pathomorphological and clinical features of myeloproliferative neoplasms (MPN) and myelodysplastic syndromes (MDS). The presence of mutations TET2, SRSF2, ASXL1, and SETBP1 in genes often associated with CMML. This leukemia is associated with persistent peripheral blood monocytes >1×109/L and >10% of WBC, does not meet WHO criteria for BCR-ABL1 CML, PMF, PV, or ET,<20% blasts in the blood and bone marrow and Dysplasia in 1 or more myeloid lineages. Monocytosis is persistent (>3 months) and all non-neoplastic causes of monocytosis have been excluded. CMML is divided into two categories: a) CMML-1: blasts (including promonocytes) lower than 5% in the peripheral blood, and lower than 10% in the bone marrow and (b) CMML‐2 blasts 5-19% in the peripheral blood, and 10-19% in the bone marrow or Auer rods are present. The 2016 revision of WHO classification has recommended distinction between MP-CMML (WBC count ≥13×109/L) and the MD- CMML (WBC <13×109/L). The difference in survival between the two subtypes is probably due to the higher prevalence of leukocytosis/monocytosis and of ASXL1 mutations in MP-CMML However, this sub classification might not provide additional prognostic value [1, 20].
Juvenile Myelomonocytic Leukemia (JMML)
Juvenile myelomonocytic leukemia often happens at an early age, and other diagnoses should be considered if the patient is over the age of six. This malignancy is associated with an increase in the number of monocytes > 1×10 9, blasts less than 20% in bone marrow and peripheral blood, BCR-ABL negative, increased hemoglobin F, increased sensitivity to GM-CSF in vitro and the presence of mutations PTPN11, KRAS, NRAS, CBL or NF1 that involve RAS signaling pathway or anomalies of the monosomy 7 [21, 22].The 2016 classification has been refined by the addition of hyper phosphorylation of STAT-5. Almost all JMML patients respond to GM-CSF indicating activation of STAT 5. This response is seen exclusively in JMML compared to healthy controls and other pediatric MPNs. So it is an important diagnostic tool [23].
Diagnostic Criteria for Atypical CML (aCML), BCR-ABL1-
By 2016 WHO classification aCML is a rare subdivision of MDS/MPN. It is now better defined in terms of molecularity and can be separated from the CNL cases, which are usually characterized by neutrophilia and which are part of the MPN family. However, the CNL is related to the presence of the CSF3R mutation, and this finding in aCML is very rare (<10%). While the aCML is related to the SETBP1 or ETNK1 mutation (in more than a third of aCML cases) and the mutations that are seen in MPN, (such as JAK2; CALR; MPL) are not in the aCML [1].
AML with Recurrent Cytogenetic Abnormalities
The cytogenetic changes in blood malignancy were a considerable revolution in diagnosis and treatment of the mentioned disease because it indicated a new way for researchers and physician of this field. There was not much change in the classification of leukemia based on cytogenesis between 2008 and 2016.
AML with t (8; 21) (q22; q22); RUNX1RUNX1T1
The t(8; 21) results in fusion of RUNX1 gene located on chromosome 21 and RUNX1T1 gene on chromosome 8 that encodes the CBFA2T1 protein. The criteria for this AML is unusual, and differ from other AML, in biological characteristics [24]. According to WHO criteria blast count is 20% or more in bone marrow or blood, except in the patients that have AML with t(15;17), t(8;21), inv(16) or t(16;16). Myeloblasts, Promyelocyte, monoblasts, promonocyte, and megakaryoblasts are considered in blast counting [25]. Patients with t (8:21) are considered to be at favorable risk for complete recovery and survival [26]. These patients express the T lymphoid marker CD19 and less CD7. They may also express the CD56 marker [25]. The determination of minimal residual disease (MRD) in AML patients with t (8; 21) may be done with specific chromosomal rearrangement, multiparametric flow cytometry (FC) and real time RT-PCR (RQ-PCR) [27]. This translocation has not changed in accordance with Table 4 in the 2008 and 2016 WHO classifications.
| 2008 | 2016 | ||||
|---|---|---|---|---|---|
| AML with t(8;21)(q22;q22);RUNX1RUNX1T1 | AML with t(8;21)(q22;q22);RUNX1RUNX1T1 | ||||
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB- MYH11 | AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB- MYH111 | ||||
| APL with t(15;17)(q22;q12); PML-RARA | APL With PML-RARA | ||||
| AML with t(9;11)(p22;q23); MLLT3-MLL | AML with t(9;11)(p22;q23); MLLT3-KMT2A | ||||
| AML with t(6;9)(p23;q34); DEK-NUP214 | AML with t(6;9)(p23;q34); DEK-NUP214 | ||||
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1- EVI1 | AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2);GATA2;MECOM | ||||
| AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15- MKL1 | AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL | ||||
| Provisional entity: AML with mutated NPM1 | AML with mutated NPM1 | ||||
| Provisional entity: AML with mutated CEBPA | AML with biallelic mutated of CEBPA | ||||
| Provisional entity:AML with BCR-ABL1 | |||||
| Provisional entity:AML with mutated RUNX1 | |||||
| Therapy-related myeloid neoplasms | Therapy-related myeloid neoplasms | ||||
| Myelodysplasia related change | Myelodysplasia related change |
Table 5: Comparison AML classification in 2008 and 2016.
AML with inv (16) (p13.1q22) or t (16; 16) (p13.1; q22); CBFB-MYH11
At the molecular level, inv (16) leads to the fusion of the core-binding factor B (CBFB, PEBP2B) gene on chromosomal band 16q22 with the smooth muscle myosin heavy chain (MYH11) gene on 16p13. On cytogenetic analysis inv (16) has been seen in 8% of adult AML patients [28]. Patients with inv (16) have an approximately favorable outcome, especially when treated with high-dose cytarabine-based consolidation regimens [29]. It is usually associated with acute myelomonocytic leukemia with eosinophilia (AML M4-Eo by the French American British classification) [30]. This translocation also has not changed in accordance with Table 4 in the 2008 and 2016 WHO classifications.
APL with PML-RARA
The PML/RARA oncoprotein induces acute promyelocytic leukemia (APL) through homo- and heterodimerization with RXRA [31]. The PML gene encodes the tumor suppressor protein PML, and at (15; 17) translocation results in a PML-RARα fusion protein which is found in most cases of APL [32] (more than 98%) and a limited number of patients without phenotypes of t (15; 17) which have different types of X-RARA-fusion [33]. Acute promyelocytic leukemia (APL) is pathologically characterized by the accumulation of promyelocytic blast cells in the bone marrow and blood [34]. This abnormality was previously known as APL with t (15; 17) (q24.1; q21.2) (previously called AML-M3). But according to Table 4, the classification for 2016 has changed the new updated names in order to emphasize the significance of the PML-RARA fusion; which may result from complex cytogenetic rearrangement other than t(15;17) (q24.1;q21.12). APL with this fusion is renamed APL with PML-RARA [1]. The RQ-PCR is used to identify this fusion [35]. All-trans retinoic acid (ATRA), one of two medicines, is given to treat the disease that communicates with the ligand binding domain present on the RARA moiety of the chimeric oncoprotein and causes both its transcriptional activation; as a result, its protolithic property disappears and leads to the differentiation of granulocytes in PML cells. Arsenic trioxide (ATO) also has an effective effect on treatment of PML. The simultaneous use of both medicines will increase their ability to cure APL. Thus, the ability of ATRA and ATO to eliminate PML/RARA a critical parameter for the success of the APL treatments is considered [36, 37]. But some patients with X-RARA-
fusion are resistant to ATRA and AS2O3 treatment [33]. For example, in t (11; 17) zinc finger protein PLZF and RARA infuse together and the morphology of the blasts and response to retinoic acid are different with t (15; 17) [38]. Therefore, in order to implemente an appropriate therapeutic method in leukemia patients with AML-M3 morphology if they are negative for t (15:17), other related mutations with gene fusion RARA are considered.
AML with t (9; 11) (p22; q23); MLLT3- KMT2A
As indicated in Table 4, due to the new revision of WHO classification the name of MLL has changed to KMT2A which was previously known as ALL-1, MLL1[1]At the molecular level, this transplantation includes the KMT2A gene in 11q23 and the MLLT3 (mixed-lineage leukemia translocate to 3) gene in 9p21, resulting in the KMT2A-MLLT3 fusion gene product. The prognosis of this translocation is controversial. According to some reports this anomaly has a good prognosis, while some do not confirm this [39].
AML with t (6; 9) (p23; q34); DEK-NUP214
Acute myelogenous leukemia (AML) with translocation involving breaks at band 23 of the short arm of chromosome 6 and band 34 of the long arm of chromosome 9, t (6; 9) (p23; q34) [40]. In 1% of cases AML [41]. Patients with this translocation give a poor response to chemotherapy, and the rate of relapse after healing is high [42]. This translocation was recognized in 1976. Usually the t(6;9) presents as de novo AML, morphologically associated with FAB type M2, bone marrow (BM) basophilia, Auer rods, and dysplasia [43]. Immunophenotype analysis is usually positive for CD13, CD33, CD34, CD38, CD45, HLA-DR, and TdT [44]. This translocation also has not changed according to Table 4 in the 2008 and 2016 WHO classifications.
AML with inv (3) (q21.3q26.2) or t (3; 3) (q21.3; q26.2); GATA2; MECOM
The subtypes acute myeloid leukemia (AML) with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); EVI1 protein are involved in regulation of transcription factors critical for hematopoiesis (eg, GATA 1, GATA2) and in epigenetic regulation [45] RPN1-EVI1 in the WHO 2008 classification is now renamed as inv(3) (q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2;MECOM in the WHO 2016 classification according to Table 4. Because the inv (3) (q21.3q26.2) or t (3; 3) (q21.3; q26.2) was not found to represent a fusion gene, but for changing position a distal GATA2 enhancer results in the overexpression of MECOM (EVI1) and functional acute failure of GATA2 [46]. Deregulated EVI1 expression is associated with low Complete Ratio rate and inferior survival; this effect is most pronounced in cytogenetic intermediate-risk AML. Allogeneic HSCT may improve survival of AML with high EVI1 [45]. Overexpression of EVI1 is caused by the inappropriate transcriptional control of the ectopic GATA2 regulatory element while GATA2 transcriptional impairment results from the removal of that same enhancer from its genomic origin. Thus it has been accepted that not RPN1, but rather the GATA2 locus is the source of the ectopic enhancer activating EVI1 in leukemia concealed inv (3)/t (3). This abnormality is seen in de novo AML and in therapy-related MDS/AML [47].
AML (megakaryoblastic) with t (1; 22) (p13; q13); RBM15-MKL1
The mutual translocation t (1; 22) (p13; q13) involving the RBM15 and MKL1 genes is an unusual abnormality that occurs under the action sets with a mutagenesis of the megakaryocytic category (AML- megakaryoblastic or AMKL) [48] which is characterized by abnormal megakaryoblasts that express platelet- specific surface glycoprotein. Bone marrow biopsy shows intense myelofibrosis, which is often difficult to aspiration of these patients [49]. This abnormality includes less than 1% of acute myeloid leukemia, but it has a higher prevalence in children. Patients with this abnormality frequently present anemia, thrombocytopenia, and organomegaly, especially hepatosplenomegaly [50] which are immunophenotypically positive for CD41, CD42b, and CD61 [51]. The prognosis of AMKL is usually bad when compared to other AML types, but AMKL with t (1; 22) (p13; q13) appears to have a better prognosis than AMKL without this cytogenetic change [50]. This translocation also has not changed in accordance with Table 4 in the 2008 and 2016 WHO classifications.
AML with mutated NPM1 [nucleophosmin (nucleolar phosphoprotein B23, numatrin)]
The most common NPM1 mutation is a 4–base pair duplication, 956dupTCTG in exon 12 (called type A), that causes a shift in the reading frame leading to replacement of the last 7 amino acids by 11 different ones in the C- terminal portion of nucleophosmin [52]. Pleiotropic functions mutations lead to abnormal cytoplasmic localization of the protein that can be diagnosed by immunohistochemistry on bone marrow sections mutations found in 25%-35% of AML [46]. Mutations of NPM1, in t(2;5)(p23;q35) in anaplastic large-cell lymphoma, t(3;5)(q25;q35) in AML, and t(5;17)(q35;q21) in acute promyelocytic leukemia are reported [52]. Leukemic blasts show high CD33 and absent to low CD34 expression associated with myelomonocytic or monocytic morphology with higher WBC and platelet counts and increased bone marrow blast percentages, higher prevalence in female sex. In younger adults with mutated NPM1 without FLT3-ITD, standard induction therapy followed by repetitive cycles of HiDAC is a reasonable treatment option; patients may not benefit from allogeneic HSCT in first complete remission. Favorable impact of NPM1 mutations in older patients is observed. Older patients with NPM1-mutated AML may benefit from intensive conventional chemotherapy. NPM1 mutations are associated with up regulation of miR-10a, miR-10b, and miR-196a, all of which reside in the genomic cluster of HOX genes that is found to be consistently overexpressed in this molecular subset [46]. Furthermore AML with mutated NPM1 shows a favorable prognosis in the absence of fms-related tyrosine kinase 3–internal tandem duplication (FLT3-ITD) mutation. For this reason, as indicated in Table 4, for AML with mutated NPM1 the 2008 WHO classification has been classified as a new category and has not changed in the 2016 version of WHO [53].
AML with Biallelic Mutations of CEBPA
The CEBP gene is located on chromosome band 19q13.1. The intron is low and rich in GC (approximately 75% containing GC) and encodes a protein that belongs to basic leucine zipper family of transcription factor. It is expressed in the cells of the myelomonocytic cell and specifically up regulated during granulocyte differentiation [54]. The finding that the improved prognosis associated with AML with mutated CEBPA, which is seen in 5% to14% of AML, is associated with biallelic, but not single, mutations of the gene which resulted in a change in that disease definition to require biallelic mutations (Table 4). Most AMLs with CEBPA mutations simultaneously carry 2 mutations (CEBPA double- mutation), usually biallelic (Nonsense mutations affecting the N-terminal region result in a truncated CEBPA isoform with dominant-negative properties, and in-frame mutations in the C-terminal basic region leucine zipper domain result in CEBPA proteins with decreased DNA binding or dimerization activity [46], whereas single heterozygous mutations (CEBPA single mutation) are less frequently seen. CEBPA double-mutation is associated with a unique gene expression profile [1]. Double mutant/biallelic cases (CEBPA dm) predict a favorable prognosis– low frequency of other mutations or other cytogenetic abnormalities that impair hematopoietic differentiation. CEBPA mutations are predominantly found in CN-AML and in cases with 9q deletion [46]. Presence of NPM1 and biallelic CEBPA trump morphological evidence of multilineage dysplasia when a mutation of NPM1 or biallelic mutation of CEBPA is present [47]. CEBPA mutations have been associated with an up regulation of members of the miR-181 family in CN- AML. Therapeutic recommendations are standard induction chemotherapy followed by three to four cycles of high-dose cytarabine. Also, AML with double CEBPA mutations may not benefit from allogeneic HSCT. In younger adults, standard induction therapy followed by repetitive cycles of HiDAC is a reasonable treatment option for AML with CEBPA mutation [46].
AML with BCR-ABL
As indicated in Table 4 AML with BCR-ABL1 is added to the new category of the 2016 WHO classification. This is rare in de novo AML cases, approximately 0.5-3 % [55]. Although the diagnostic distinction between de novo AML with BCR-ABL1 and blast transformation of CML may be difficult without adequate clinical information, in recurrent not universal deletion of antigen receptor genes (IGH, TCR), IKZF1 and/or CDNK1A gene may support a diagnosis of de novo disease versus by Blasting Phase of CML[1]. This criteria, suggested for differentiating Philadelphia chromosome (Ph+) AML from CML-MBC (CML in myeloid blastic crisis) includes an absence of a clinical history of a hematologic malignancy, lack of evidence of chronic phase or accelerated phase of CML and lack of clinical and laboratory features of CML such as splenomegaly and basophilia [56]. In CML-MBC, the most common additional abnormalities are trisomy 8, trisomy 19 or is chromosome 17q, in addition to t(9;22) [55]. These findings support the identification of AML with BCR-ABL1 as a distinct entity from CML myeloid blast crisis. A combination of AML chemotherapy and continuing Imatinib, can provide recovery and a further allogeneic stem cell transplant (ASCT) is the best stabilizing therapy for the patient with Ph+ AML [57].
AML with Mutated RUNX1
As seen in Table 4, a provisional category of AML with mutated RUNX1 (runt-related transcription factor 1) is a myeloid transcription factor described as recurrently mutated in de novo acute myeloid leukemia (AML; ~ 10%) [58]. The RUNX1 gene is located at chromosomal band 21q22. It contains a “Runt homology domain” (RHD) that is responsible for heterodimerization with the core- binding factor (or PEBP2) as well as transcription factor and for DNA binding. The second important part of the RUNX1 protein is the transactivation domain followed by a 5-amino acid sequence of the gene products [59]. This gene is required for definitive hematopoiesis, and its functional mutation leads to leukemia [60]. Multivariate Cox regression analysis defends RUNX1 as a new entity in the AML without taking the dysplasia characteristics into consideration [58]. RUNX1 mutations have been associated with undifferentiated (M0) morphology and with specific chromosome aberrations, such as trisomy 21 and trisomy 13 [46]. This new provisional disease category appears to represent a biologically distinct group with a possibly worse prognosis than other AML types [1].
AML with Mutated C-KIT
C-KIT and its ligand (stem cell factor) play a key role in the survival, proliferation, differentiation and activation of hematopoietic progenitors. Proto-oncogene C-KIT is expressed in 80% of cases of AML, which represents a poor prognosis. Identifying the c-kit makes treatment more specific and appropriate [61, 62].
AML with Mutated Nup
The NPC is one of the largest and most complex proteins in the cell, composed of about 30 proteins in multiple copies. The task is to set up a nuclear- cytoplasmic transport of macromolecules larger than 40 KD. NPC consists of nucleoproteins. Many AML, N- terminal Nup98 proteins have been displaced with the C- terminal of 30 different partners. But how this mechanism works is not well known [63, 64].
AML with Mutated FLT3
Mutations in the fms-related tyrosine kinase 3 (FLT3) gene are found in many AML subtypes. In up to 30% of acute myeloid leukemia (AML) patients [67]. In general, FLT3 mutations can be divided into 2 categories: 1- internal tandem duplications (FLT3/ITD mutations) in or near the juxtamembrane domain of the receptor and 2- point mutations resulting in single amino acid exchange occurring within the activation loop of the tyrosine kinase domain (FLT3/TKD mutations) [65]. Internal tandem duplication mutations of the FMS-like tyrosine kinase-3 receptor (FLT3/ITD mutations) are one of the most common molecular abnormalities found in de novo acute myeloid leukemia (AML) [68]. Approximately 20–30% of AML patients carry an internal tandem duplication (ITD) mutation in the FLT3 gene but mutations in the tyrosine kinase domain (TKD) of FLT3 are less frequent (7%) and currently have no clinically significant impact [69]. FLT3 appertain to the class III receptor tyrosine kinase (RTK) family, including FMS, c-KIT, platelet derived growth factor receptor, and platelet-derived growth factor receptor β [70]. FLT3 plays an important role in hematopoiesis by regulating proliferation, differentiation, and apoptosis of cell progenitors [71]. The prognosis for patients with FLT3-ITD mutations in the juxtamembrane domain, is worse than the patients with wild-type FLT3 when treated with standard therapy [72]. Allogeneic HSCT should be considered in AML with FLT3-ITD. FLT3- ITD has been reported to be associated with miR-155 up regulation [46].
Acute Myeloid Leukemia, Not Otherwise Specified (NOS)
As seen in Table 5, [74] in 2016 WHO classification AML with not otherwise specified, (NOS) has only a single change: The subgroup of acute erythroid leukemia and the type of erythroid/myeloid (formerly as cases of ≥50% erythroid precursors in bone marrow and ≥20% of myeloblasts in non-erythroid cells) has been excluded from the classification of the AML. In the new classification, myeloblast is always measured as a percentage of the total cells in the bone marrow. And in most cases, with <20% of the total blast cells, is now classified as MDS (usually MDS with excess blast). These changes are based on the biological relationships of the erythroid/myeloid type of acute erythroid leukemia to MSD in terms of clinical findings, morphological features, genetic abnormalities, and attempts to align the expression of blast percentages in all myeloid neoplasms [1, 47]. Items with ≥50% erythroid cells and ≥20% of myeloblasts (previously known as M6 in the FAB (French- American-British) category) should now be diagnosed if they have criteria for AML with myelodysplasia-related changes. Therefore, in the new classification of WHO, dysplasia alone can be sufficient for the classification of AML with myelodysplasia-related changes. And items with ≥20% of the myeloblast that do not have AML with myelodysplasia-related changes classification criteria should be included in AML; NOS [65].
AML Associate with Down Syndrome
The risk of AML in patients with Down syndrome is increased. Myeloid proliferations related to Down syndrome include transient abnormal myelopoiesis (TAM) and myeloid leukemia associated with Down syndrome About [75, 76]. 20% to 30% of children with myeloid leukemia had a previous history of TAM [77]. The myeloid leukemia and TAM blasts cannot be differentiated in terms of phenotypic and morphologically. In this group of malignancies blasts showed megakaryocytic differentiation with aberrant CD7 expression [78]. The mutation in GATA-1 is seen [79]. The mutation in JAK-STAT signaling pathway has been added in the 2016 classification. Because this mutation is seen only in myeloid leukemia associated with Down syndrome, while a mutation in GATA-1 is seen in TAM and myeloid leukemia. There is also a small number of leukemia cells that are unrelated to Down-syndrome that have GATA-1 mutation. Therefore, JAK-STAT reduces the probability of error in the diagnosis [77].
AML associate with Sarcoma
Myeloid sarcoma is a tumor lesion of immature cells of the granulocytic series that can be created anywhere in the body. Most of the tumors can be seen in soft tissues, bones, lymph nodes, peritoneum, gastrointestinal tract, etc. [80]. This tumor lesion can occur either in the form of denevo or at the same time as AML, MDS and MPD. It can be a relapse symptom of the disease. Isolated into a sarcoma in which the bone marrow is not involved. Usually sarcoma of bone marrow involvement is associated with leukemia [79]. In the 2016 classification, it is proposed to classify cases that are not associated with bone marrow involvement in a separate subgroup, since treatment for those with bone marrow involvement is different and more difficult [81].
AML Associate with Ambiguous Lineage
Leukemia with ambiguous lineage that does not express specific marker of the lymphoid either myeloid lineage, or the specific marker of both classes is expressed on blast cells [82]. In 2016, it is better to classify this category of leukemia based on genetic variation. Because it makes it more specific and more suitable for treatment [83].
Natural Killer (NK) Cell Lymphoblastic Leukemia/Lymphoma
This leukemia expresses markers CD16, CD56 and is negative for CD3. If CD123 is positive, it has dendritic origin [84]. This leukemia in the 2008 classification, according to the Table 6 was a subset of the ambiguous leukemia. Because of this, there are few specific markers that are hard to distinguish from acute myeloid leukemia. CD56 is a sensitive marker for NK detection, but not specific. Diagnosis of this malignancy is possible at a time when other leukemia is excelled [83]. In the 2016 classification, it is located in the LGL lymphoid subgroup [85] (Table 6).
- 2008
- 2016
- Acute undifferentiated
- Acute undifferentiated leukemia leukemia
- Mixed phenotype acute
- Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCRleukemia with t(9;22)(q34;q11.2); BCR-
- ABL1
- ABL1
- Mixed phenotype acute leukemia with t(v;11q23);
- Mixed phenotype acute leukemia with t(v;11q23);
- MLL rearranged
- MLL rearranged
- Mixed phenotype acute leukemia, B-myeloid, NOS
- Mixed phenotype acute leukemia, B-myeloid, NOS
- Mixed phenotype acute leukemia, T-myeloid, NOS
- Mixed phenotype acute leukemia, T-myeloid, NOS
- Natural killer (NK) cell lymphoblastic leukemia/lymphoma (Provisional entity)
Table 7: AML associated with Ambiguous lineage.
Adding molecular findings in leukemia has not only been able to solve the problems in diagnosis, but also has a therapeutic approach. The classification of leukemia by the WHO and the collaboration of researchers seems to promote the unknown aspects of mutations and molecular damages in this part of science, and it is still necessary to continue to reveal unknown diseases.
References
-
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, et al. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20): 2391-2405.
-
Pophali PA, Patnaik MM (2016) The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer J 22(1): 40-50.
-
Elliott MA, Tefferi A (2016) Chronic neutrophilic leukemia 2016: Update on diagnosis, molecular genetics, prognosis, and management. Am J Hematol 91(3): 341-349.
-
Szuber N, Tefferi A (2018) Chronic neutrophilic leukemia: new science and new diagnostic criteria. Blood cancer journal 8(2): 19.
-
Rohrabaugh S, Kesarwani M, Kincaid Z, Huber E, Leddonne J, et al. (2017) Enhanced MAPK signaling is essential for CSF3R-induced leukemia. Leukemia 31(8): 1770-1778.
-
Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi A, et al. (2016) The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev 30(6): 453-459.
-
Barbui T, Thiele J, Vannucchi A, Tefferi A (2014) Rethinking the diagnostic criteria of polycythemia vera. Leukemia 28(6): 1191-1195.
-
Griesshammer M, Gisslinger H, Mesa R (2015) Current and future treatment options for polycythemia vera. Ann Hematol 94(6): 901-910.
-
Barbui T, Thiele J, Vannucchi A, Tefferi A (2015) Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J 5(8): e337.
-
Tefferi A, Thiele J, Vannucchi A, Barbui T (2014) An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 28(7): 1407-1413.
-
de Melo CP (2016) Primary myelofibrosis: current therapeutic options. Rev Bras Hematol Hemoter 38(3): 257-263.
-
Tefferi A (2016) Primary myelofibrosis: 2017 update on diagnosis, risk‐stratification, and management. Am J Hematol 91(12): 1262-1271.
-
Clinton A, McMullin MF (2016) The Calreticulin gene and myeloproliferative neoplasms. J Clin Pathol 69(10): 841-845.
-
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, et al. (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. New England Journal of Medicine 369(25): 2379-2390.
-
Brockow K, Metcalfe DD (2010) Mastocytosis. Anaphylaxis 95: 110-124.
-
Valent P, Akin C, Hartmann K, Nilsson G, Reiter A, et al. (2017) Advances in the classification and treatment of mastocytosis: current status and outlook toward the future. Cancer Res 77(6): 1261-1270.
-
Valent P, Akin C, Metcalfe DD (2017) Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 129(11): 1420-1427.
-
Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, et al. (2018) The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J 8(2): 15.
-
Hall J, Foucar K (2010) Diagnosing myelodysplastic/myeloproliferative neoplasms: laboratory testing strategies to exclude other disorders. Int J Lab Hematol 32(6): 559-571.
-
Hamed NA (2017) Chronic Myelomonocytic Leukemia: Current Classification, Diagnosis and Risk Stratification. Cancer Therapy & & Oncology International journal of hematology 5(1): 1-4.
-
Emanuel P (2008) Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia 22(7): 1335-1342.
-
Vardiman J, Hyjek E (2011) World health organization classification, evaluation, and genetics of the myeloproliferative neoplasm variants. ASH Education Program Book 2011: 250-256.
-
Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, et al. (2013) GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood 121(25): 5068- 5077.
-
Reikvam H, Hatfield KJ, Kittang AO, Hovland R, Bruserud Ø (2011) Acute myeloid leukemia with the t (8;21) translocation: clinical consequences and biological implications. J Biomed Biotechnol 2011: 104631.
-
Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, et al. (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115(3): 453-474.
-
Micol J-B, Duployez N, Boissel N, Petit A, Geffroy S, et al. (2014) Frequent ASXL2 mutations in acute myeloid leukemia patients with t (8;21)/RUNX1- RUNX1T1 chromosomal translocations. Blood 124(9): 1445-1449.
-
Perea G, Lasa A, Aventin A, Domingo A, Villamor N, et al. (2006) Prognostic value of minimal residual disease (MRD) in acute myeloid leukemia (AML) with favorable cytogenetics [t (8; 21) and inv (16)]. Leukemia 20(1): 87-94.
-
Paschka P, Du J, Schlenk RF, Gaidzik VI, Bullinger L, et al. (2013) Secondary genetic lesions in acute myeloid leukemia with inv (16) or t (16; 16): a study of the German-Austrian AML Study Group (AMLSG). Blood 121(1): 170-177.
-
Corbacioglu A, Scholl C, Schlenk RF, Eiwen K, Du J, et al. (2010) Prognostic impact of minimal residual disease in CBFB-MYH11–positive acute myeloid leukemia. J Clin Oncol 28(23): 3724-3729.
-
Tirado CA, Valdez F, Klesse L, Karandikar NJ, Uddin N, et al. (2010) Acute myeloid leukemia with inv (16) with CBFB–MYH11, 3′CBFB deletion, variant t (9;22) with BCR-ABL1, and del (7)(q22q32) in a pediatric patient: case report and literature review. Cancer Genet Cytogenet 200(1): 54-59.
-
Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, et al. (2010) PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell 18(1): 88-98.
-
Chang HR, Munkhjargal A, Kim M-J, Park SY, Jung E, et al. (2017) The functional roles of PML nuclear bodies in genome maintenance. Mutat Res 809: 99-107.
-
Tomita A, Kiyoi H, Naoe T (2013) Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int J Hematol 97(6): 717-725.
-
Wang K, Wang P, Shi J, Zhu X, He M, et al. (2010) PML/RAR alpha targets promoter regions containing PU. 1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer cell 17(2): 186-197.
-
Ommen HB, Schnittger S, Jovanovic JV, Ommen IB, Hasle H, et al. (2010) Strikingly different molecular relapse kinetics in NPM1c, PML-RARA, RUNX1- RUNX1T1, and CBFB-MYH11 acute myeloid leukemias. Blood 115(2): 198-205.
-
Goto E, Tomita A, Hayakawa F, Atsumi A, Kiyoi H, et al. (2011) Missense mutations in PML-RARA critical for the lack of responsiveness to arsenic trioxide treatment. Blood 118(6): 1600-1609.
-
Isakson P, Bjørås M, Bøe SO, Simonsen A (2010) Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 116(13): 2324-2331.
-
Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ (1996) The t (5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood 87(3): 882-886.
-
de Matos RRC, Garcia DRN, Cifoni E, Othman MA, de Souza MT, et al. (2017) GAS6 Oncogene and Reverse MLLT3-KMT2A Duplications in an Infant with Acute Myeloid Leukemia and a Novel Complex Hyperdiploid Karyotype: Detailed High-Resolution Molecular Cytogenetic Studies. Cytogenet Genome Res 152(1): 33-37.
-
Chi Y, Lindgren V, Quigley S, Gaitonde S (2008) Acute myelogenous leukemia with t (6;9) (p23;q34) and marrow basophilia: an overview. Arch Pathol Lab Med 132(11): 1835-1837.
-
Ishiyama K, Takami A, Kanda Y, Nakao S, Hidaka M, et al. (2012) Allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with t(6; 9) (p23; q34) dramatically improves the patient prognosis: a matched-pair analysis. Leukemia 26(3): 461-464.
-
Tarlock K, Alonzo TA, Moraleda PP, Gerbing RB, Raimondi SC, et al. (2014) Acute myeloid leukaemia (AML) with t(6;9) (p23;q34) is associated with poor outcome in childhood AML regardless of FLT3‐ITD status: a report from the Children's Oncology Group. Br J Haematol 166(2): 254-259.
-
Sandahl JD, Coenen EA, Forestier E, Harbott J, Johansson B, et al. (2014) t (6; 9) (p22; q34)/DEK- NUP214 rearranged pediatric myeloid leukemia: an international study on 62 patients. Haematologica 99(5): 865-872.
-
Gupta M, Kumar JA, Sitaram U, Neeraj S, Nancy A, et al. (2010) The t (6; 9)(p22; q34) in myeloid neoplasms: a retrospective study of 16 cases. Cancer Genet Cytogenet 203(2): 297-302.
-
Marcucci G, Haferlach T, Döhner H (2011) Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol 29(5): 475- 486.
-
Gröschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, et al. (2014) A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157(2): 369-381.
-
Hong M, He G (2017) 2016 Revision to the WHO classification of acute myeloid leukemia. J Transl Int Med 5(2): 69-71.
-
Margolskee E, Saab J, Geyer JT, Aledo A, Mathew S (2017) A Novel Variant t(1;22) Translocation– ins(22;1)(q13;p13p31)–in a Child with Acute Megakaryoblastic Leukemia. Am J Case Rep 18: 422- 426.
-
Gruber TA, Downing JR (2015) The biology of pediatric acute megakaryoblastic leukemia. Blood 126(8): 943-949.
-
Torres L, Lisboa S, Vieira J, Cerveira N, Santos J, et al. (2011) Acute megakaryoblastic leukemia with a four‐way variant translocation originating the RBM15–MKL1 fusion gene. Pediatric Blood Cancer 56(5): 846-849.
-
Schweitzer J, Zimmermann M, Rasche M, von Neuhoff C, Creutzig U, et al. (2015) Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 94(8): 1327-1336.
-
Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD (2007) Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 109(2): 431-448.
-
Falini B, Macijewski K, Weiss T (2010) Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 115(18): 3776-3786.
-
Ng CWS, Kosmo B, Lee P-L, Lee CK, Guo J, et al. (2018) CEBPA mutational analysis in acute myeloid leukaemia by a laboratory-developed next-generation sequencing assay. J Clin Pathol 71(6): 522-531.
-
Reboursiere E, Chantepie S, Gac A-C, Reman O (2015) Rare but authentic Philadelphia-positive acute myeloblastic leukemia: two case reports and a literature review of characteristics, treatment and outcome. Hematol Oncol Stem Cell Ther 8(1): 28-33.
-
Gopal MM, Kotwal J, Kapoor R (2014) Acute Myeloid Leukemia with BCR/ABL Fusion Chimera. Indian J Hematol Blood Transfus 30(1): 280-282.
-
Fei X-H, Wu S-L, Sun R-J, Zhou J-R, Wang J-B, et al. (2012) Clinical analysis of 12 cases of acute myeloid leukemia with Ph chromosome and BCR-ABL positive. Zhongguo Shi Yan Xue Ye Xue Za Zhi 20(3): 545-548.
-
Haferlach T, Stengel A, Eckstein S, Perglerová K, Alpermann T, et al. (2016) The new provisional WHO entity ‘RUNX1 mutated AML’ shows specific genetics but no prognostic influence of dysplasia. Leukemia 30(10): 2109-2112.
-
Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, et al. (2011) RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 117(8): 2348-2357.
-
Tang J-L, Hou H-A, Chen C-Y, Liu C-Y, Chou W-C, et al. (2009) AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood 114(26): 5352-5361.
-
Ayatollahi H, Shajiei A, Sadeghian MH, Sheikhi M, Yazdandoust E, et al. (2017) Prognostic importance of C-Kit mutations in core binding factor acute myeloid leukemia: A systematic review. Hematol Oncol Stem Cell Ther 10(1): 1-7.
-
Heo S-K, Noh E-K, Kim JY, Jo J-C, Choi Y, et al. (2017) Radotinib induces high cytotoxicity in c-KIT positive acute myeloid leukemia cells. Eur J Pharmacol 804: 52-56.
-
Franks TM, Hetzer MW (2013) The role of Nup98 in transcription regulation in healthy and diseased cells. Trends Cell Biol 23(3): 112-117.
-
Franks TM, McCloskey A, Shokirev M, Benner C, Rathore A, et al. (2017) Nup98 recruits the Wdr82– Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev 31(22): 2222-2234.
-
Levis M (2013) FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program 2013: 220-226.
-
Devillier R, Gelsi‐Boyer V, Brecqueville M, Carbuccia N, Murati A, et al. (2012) Acute myeloid leukemia with myelodysplasia‐related changes are characterized by a specific molecular pattern with high frequency of ASXL1 mutations. Am J Hematol 87(7): 659-662.
-
Levis M, Ravandi F, Wang ES, Baer MR, Perl A, et al. (2011) Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 117(12): 3294-3301.
-
Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, et al. (2010) FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 115(7): 1425-1432.
-
El Fakih R, Rasheed W, Hawsawi Y, Alsermani M, Hassanein M (2018) Targeting FLT3 Mutations in Acute Myeloid Leukemia. Cells 7(1).
-
Kindler T, Lipka DB, Fischer T (2010) FLT3 as a therapeutic target in AML: still challenging after all these years. Blood 116(24): 5089-5102.
-
Fischer T, Stone RM, DeAngelo DJ, Galinsky I, Estey E, et al. (2010) Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 28(28): 4339.
-
Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, et al. (2009) AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114(14): 2984-2992.
-
Weinberg OK, Seetharam M, Ren L, Seo K, Ma L, et al. (2009) Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood 113(9): 1906-1908.
-
Zahid MF, Parnes A, Savani BN, Litzow MR, Hashmi SK (2016) Therapy-related myeloid neoplasms-what have we learned so far? World J Stem Cells 8(8): 231- 242.
-
Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, et al. (2008) Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood 111(3): 1575-1583.
-
Xavier AC, Ge Y, Taub JW (2009) Down syndrome and malignancies: a unique clinical relationship: a paper from the 2008 william beaumont hospital symposium on molecular pathology. J Mol Diagn 11(5): 371-380.
-
Hama A, Muramatsu H, Makishima H, Sugimoto Y, Szpurka H, et al. (2012) Molecular lesions in childhood and adult acute megakaryoblastic leukaemia. Br J Haematol 156(3): 316-325.
-
Wang SA, Hasserjian RP (2015) Acute erythroleukemias, acute megakaryoblastic leukemias, and reactive mimics: a guide to a number of perplexing entities. Am J Clin Pathol 144(1): 44-60.
-
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, et al. (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114(5): 937-951.
-
Samborska M, Derwich K, Skalska-Sadowska J, Kurzawa P, Wachowiak J (2016) Myeloid sarcoma in children–diagnostic and therapeutic difficulties. Contemp Oncol 20(6): 444-448.
-
Thiel U, Wawer A, von Luettichau I, Bender H-U, Blaeschke F, et al. (2016) Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget 7(43): 70959-70968.
-
Béné MC (2009) Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94(7): 891-893.
-
Shi R, Munker R (2015) Survival of patients with mixed phenotype acute leukemias: A large population-based study. Leuk Res 39(6): 606-616.
-
Miesner M, Haferlach C, Bacher U, Weiss T, Macijewski K, et al. (2010) Multilineage dysplasia (MLD) in acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities and a prior history of MDS or MDS/MPN but has no independent prognostic relevance: a comparison of 408 cases classified as" AML not otherwise specified"(AML-NOS) or" AML with myelodysplasia- related changes"(AML-MRC). Blood 116(15): 2742- 2751.
-
Pilichowska ME, Fleming MD, Pinkus JL, Pinkus GS (2007) CD4+/CD56+ Hematodermic Neoplasm (“Blastic Natural Killer Cell Lymphoma”) Neoplastic Cells Express the Immature Dendritic Cell Marker BDCA-2 and Produce Interferon. Am J Clin Pathol 128(3): 445-453.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies