Compound Heterozygous Sickle Cell and Beta Thalessemia Trait with High Hb F: A Rare Case from India
Hemoglobinopathies constitute a good number of percentage of cases causing hemolytic anaemia. Hemoglobin is a tetramer consisting of 2 globin chains. Any defect is these chains cause abnormal hemoglobin synthesis. We hereby report a rare case of compound Sickle Beta Thalassemia with high fetal Hemoglobin. The high percentage of Hb F makes it even a more rare case which has not been reported in literature so far.
Introduction
Hemolytic anemia is a major public health problem in various countries including India. Hemoglobinopathies constitute an important cause of hemolytic anemia. According to WHO reports, nearly 7% of global population carries an abnormal hemoglobin gene [1]. About 300,000-500,000 are born annually with significant hemoglobin disorders. They consist of two major groups-Thalassemia and Sickle cell syndromes. In India, β-Thalassemia is prevalent across the country, with an average frequency of carriers being 3-4%. A higher frequency has been observed in certain communities, such as Sindhis, Punjabis, Gujaratis, Bengalis, Mahars, Kolis, Saraswats, Lohanas and Gaurs. HbS is highly prevalent in the tribal populations of Southern, Central and Western states reaching as high as 48% in some communities [2]. A rare entity called compound heterozygous comprises of Sickle Beta Thalassemia which constitute 0.02% of overall prevalence in India is seen with highest prevalence in Bangalore (0.06%) [3].
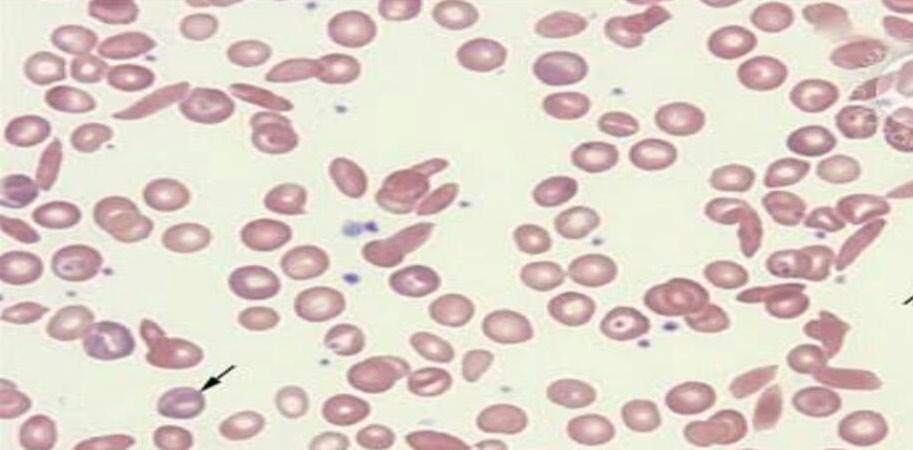
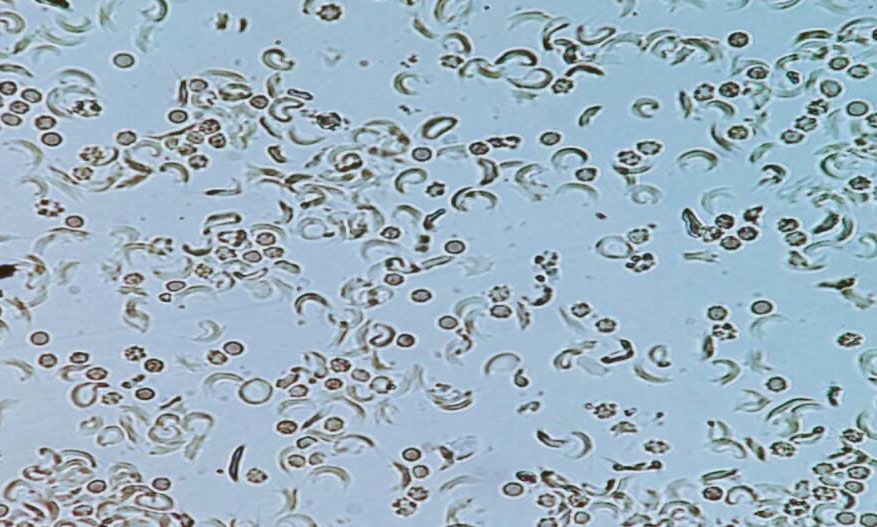
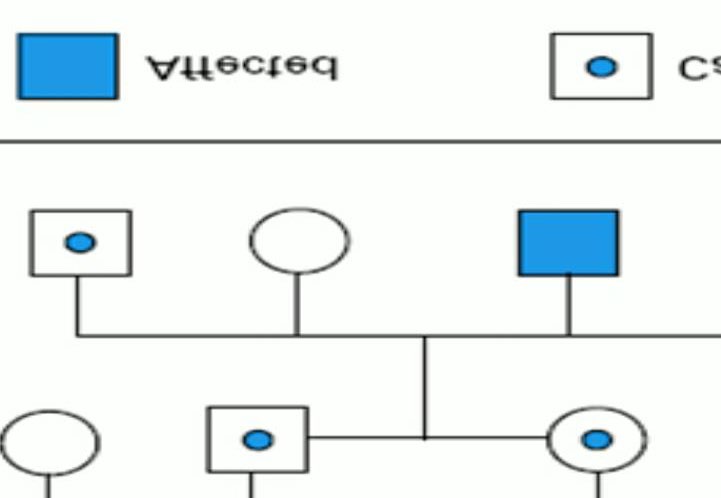
A 12yr old boy presented to pediatric OPD with low grade fever (100.5F). On General examination, patient was average built. Pallor was present. No icterus and cyanosis. He had past history of jaundice at the age of 4 years and 3 episodes of respiratory infections since birth. On systemic examination, splenomegaly was present. However liver was not palpable. Peripheral blood smear showed microcytic hypochromic RBCs, few sickled cells and some target cells (Figure 1). Hemoglobin was low 6.4gm%. RBC count was 2.85x 106/μl, MCV was 66.1fl, MCH was 21.1pg, MCHC was 31.9g/dl. Reticulocyte count was 5.5%. Further laboratory investigations were advised which showed high LDH level of 1733U/L, S.Ferritin was normal (147ng/ml). Vitamin B12 & Folate were within normal limits. G6PD showed normal activity. DCT &ICT were Negative. In view of anemia, splenomegaly, raised LDH and negative coombs & G6PD test, a sickle cell preparation was made which showed sickled RBCs (Figure 2). HPLC was done in which HbF was 35.1%, HbA was 5.2%, HbA2 was 4.2%, S-window was 53.5% (Figure 3). Blood transfusion was advised immediately. Considering the unusual presentation the complete investigation of entire family was advised which showed that both parents were carrier of thalassemia trait and sickle cell trait. Even the siblings of patient were carrier (Figure 4).
Complete blood picture of patient’s father revealed Hb was 11.3gm%, RBC 5.5 X106/μl, MCV was 62.8fl, MCH was 20.4pg, WBC & platelets were normal. Peripheral blood film showed microcytic hypochromic RBCs, anisopoikilocytosis and many target cells. However sickle cell preparation was negative. HPLC was done which showed HbF was 1.9% HbA was 81.4%, HbA2 was 6.2%. A diagnosis of β-Thalassemia trait was made for father. Similarly blood picture of patient’s mother revealed Hb was 12.2 gm%, RBC was 4.7X106/ μl, MCV was 75.8fl, MCH was 25.8pg. Peripheral blood film showed microcytosis, hypochromia and many sickle shaped red blood cells. Sickling test was positive. HPLC was done which showed HbF 0.3%, HbA was 53.4%, HbA2 was 3.5%, S-window was 35.9%. A diagnosis of sickle cell trait was made for mother.
Patient’s younger brother was also investigated and as predicted he came out to be sickle cell trait as well. His blood picture revealed microcytosis, hypochromia, and occasional Sickle Cells. Sickling test was positive. HPLC was done which showed HbF 5.0% HbA 50%, HbA2 3.1%, S-window 34.5%. Since the investigation were done outside our hospital so no graph available. Solubility test was done in all family members which was positive in patient, his brother and mother. Geographical history of origin was asked which revealed father was from Puducherry and mother from Vishakapatnam. HPLC graph of family was not available. However, an interesting finding here was that in all the family members HbF was within normal levels, but in our patient HbF was high- 35.1% which makes it a rare entity. Considering the family picture, a diagnosis of double heterozygous sickle cell and beta thalessemia with high HbF was made. Although no history of consangious marriage was given by parents, but they were counselled regarding the disease and further precautions to be taken.

Discussion
Hemoglobinopathies constitute an important cause of hemolytic anaemia. Beta-thalassemia and Sickle Cell Disease create a major burden of morbidity and mortality. Sickle Cell Disease results from a single amino acid substitution in the gene encoding the b-globin subunit (B6 glu>val), that produces the abnormal hemoglobin named HbS. The combination of the Sickle Cell mutation and Beta Thalassemia mutation give rise to a compound heterozygous condition known as HbS/Beta Thalassemia, which was first reported by Dacie J [4]. A spectrum of β thalassemia mutation is associated with Sickle Cell disease, resulting in difference in quantity of Beta globin synthesis and amount of HbA2. Usually it’s classified into HbS/β0 and HbS/β+ Thalassemia. HbS/ β+ Thalassemia patients have a higher hemoglobin level and lower reticulocyte count than those of HbS/β0 thalessemia patients [5]. HbS/ β+ Thalassemia can further be classified into Type 1 (HbA - 1-7%), Type 2(HbA- 7-14%) and Type 3 (HbA-14-25%) depending on the level of HbA [4, 6]. But the sub-classification of HbS/β+ Thalassemia is less accepted and has been considered of low utilility.
Various studies have shown that the clinical course of Sickle Cell Disease/ β0 Thalassemia is generally less severe, than that of homozygous Sickle Cell disease [7]. In our case the patient presented with milder form of clinical course. A probable explanation could be that our patient had high percentage of HbA2 & HbF which has an inhibitory effect on sickling process as they don’t participate in the tactoid formation and dilute HbS, which inhibit the polymerization. The effect of thalassemia results into inducing microcytosis and hypochromia in sickle cells which improves the circulatory competence and reduce hemolysis [8].
Blood picture of patient (in the present case) is similar to those in homozygous Sickle cell disease with microcytosis, hypochromia, target cells and Sickle Cells which makes it difficult to distinguish from homozygous state. Only HPLC seems to be an aid with the support of evidence inheritance analysis. Thein, et al. described an Asian Indian family with a non-deletion form of hereditary persistence of fetal haemoglobin (HPFH) and β0 thalassemia. The patient was homozygous beta zero thalassemia had an unusually mild form of the disease, which was attributed to the coinheritance of HPFH [9]. However in our case the patient is compound heterozygous sickle and thalassemia trait which in itself is rare and very few case reports are found [10]. But the most interesting finding is the high Hb F level in our case which makes it extremely rare and is reported first time in literature from India. There is coinheritance of other condition called hereditary persistence of fetal hemoglobin which has modified the whole spectrum of disease. It has been documented in some studies that conditions like HPFH, iron deficiency anaemia, alpha thalassemia may change the overall laboratory findings and symptoms in patients [11]. Molecular studies were advised from outside laboratories to the family, as it was not available in our institute. Parents were not able to get it done from outside laboratories due to financial constraints. The child was managed with packed red blood cell transfusions and symptomatic relief was given. The general condition improved considerably till the next follow up.
Conclusion
Hemoglobinopathies can be a diagnostic challenge to physician as well as hematologist. Differentiation of Sickle cell Anemia & Sickle Beta Thalassemia syndromes has to be done carefully due to close similarity of symptoms and laboratory features. Early and correct diagnosis can enhance the quality of life of such patient. This case underlines the importance of a through history, clinical examination and appropriate laboratory testing for suitable management and prognostication as well as genetic counseling. Parental studies proved crucial in confirming the double heterozygous state of sickle cell disease/thalassemia especially when there is limitation of genetic studies.
References
-
WHO (2008) Management of Haemoglobin Disorders. Report of Joint WHO-TIF Meeting on the Management of Haemoglobin Disorders. Nicosia, Cyprus, 16-18 November 2007, World Health Organization pp: 1-2.
-
National Health Mission Guidelines on Hemoglobinopathies in India Prevention and control of hemoglobinopathies in India-Thalassemia, Sickle cell disease and other variant hemoglobins. Ministry of Health and Family Welfare, Govt of India, 2016.
-
Jegan NK, Ramcharan R, Udayashankar D, Durga K, Rajasekaran D (2014) Sickle Beta thalassemia. A case report. Chettinad Health City Medical Journal 3(3): 133- 135.
-
Dacie J (1988) The hereditary hemolytic anaemias. 3rd (Edn.), New York: Churchill Livingstone.
-
Hankins JS, Wang WC (2014) Sickle cell anaemia and other sickling syndromes. Wintrobe’s Clinical Hematology 1(13): 823-859.
-
Giordano PC (2013) Strategies for basic laboratory diagnostics of the haemoglobinopathies in multiethnic societies: interpretation of results and pitfalls. Int J of Lab Haematol 35(5): 465-479.
-
Serjeant GR, Serjeant BE, Fraser RA, Hambleton IR, Higgs DR, et al. (2011) Hb S-betathalassemia: molecular, hematological and clinical comparisons. Hemoglobin 35(1): 1-12.
-
Fintel R, Schwyzer R, Poole J, Ali N (2013) Compound heterozygous sickle cell disease and β0-thalassaemia: An interesting case. S Afr J CH 7(2): 70-73.
-
Thein SL, Weatherall DJ (1989) A non-deletion hereditary persistence of fetal hemoglobin (HPFH) determinant not linked to the beta-globin gene complex. Prog Clin Biol Res 316: 97-111.
-
Shreshtha B, Karmacharya K, Singh J, Kotwal J, Devgan A (2011) Compound Heterozygous Sickle and Thalassemia Trait: A Case Report J Nep PaedtrSoc 31(2): 130-133.
-
Corrina M (2006) Sickle cell disease. In: Arceri R, et al. (Eds.), Pediatric hematology. 3rd (Edn.), Massachusetts: Blackwell Publishings, pp: 213-226.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies