Contribution of Xmn I Polymorphism in the Variation of Hemoglobin F in Thalassemia Intermedia Patients in Pakistan
Background: The −158 (C→T) nucleotide change, known as Xmn I polymorphism, occurs in Gγ-globin gene promoter and may result in elevated fetal hemoglobin (HbF). However some studies did not find any association of HbF with the mutation. This study was taken to confirm the influence of Xmn-I polymorphism on HbF production in thalassemia intermedia patients in Pakistani population. Methods: Blood samples from 100 known thalassemia intermedia patients were collected and analyzed for Xmn-I polymorphism and levels of hemoglobin F. Results: High levels of HbF were found in Thalassemia intermedia patients being heterozygous and homozygous for Xmn-I polymorphism. Conclusion: XmnI polymorphism (C-T) of the Gγ-globin gene promoter is associated with increased expression of the Gγ-globin gene, higher production of HbF and lesser clinical severity.
Introduction
The−158 (C→T) nucleotide change, known as Xmn I polymorphism, occurs in Gγ-globin gene promoter and may results in elevated fetal hemoglobin (HbF). Role of XmnI polymorphism in the phenotype and levels of HbF in homozygous and compound heterozygous β‑thalassemia has been in research in different populations [1, 2]. Higher levels of HbF in beta-thalassemia with the XmnI polymorphism shows the influence of this site on the gene expression of β-globin [3]. However some studies did not find any association between the mutation Ggamma -158 C→T and Hb F content in beta thalassemia intermedia patients [4]. This study was taken to confirm the influence of Xmn-I polymorphism on HbF production in thalassemia intermedia patients in Pakistani population.
Materials and Methods
Xmn-I can recognize the C-T polymorphism at position – 158 from the Cap site of the G β-globin gene [5]. In order to demonstrate this polymorphism, a 641bp fragment of DNA flanking the polymorphism was amplified using the following primers. 5’ – GAA CTT AAG AGA TAA TGG CCT AA 5’ – ATG ACC CAT GGC GTC TGG ACT AG The reaction mixture was prepared by adding 20 µl of buffer, 1 µl of primer, 2 µl of DNA and 0.1 µl of Taq. The PCR conditions were for the RFLP protocol with 94oC for 1 minute denaturation time, Annealing at 60°C for 1 minute extension at 72°C for 1 minute and 3 minutes of final extension at 72°C. Numbers of cycles carried out were 30. Tubes were removed from the PCR machine and the amplified fragment was digested with 10 units of enzyme Pdm-I (Fermentus) at 37°C overnight and the results were recorded after electrophoresis on 6% Acrylamide gel (Figures 1 & 2).

The presence of different hemoglobins including hemoglobin A, A2 and F was carried out by cellulose acetate gel electrophoresis.
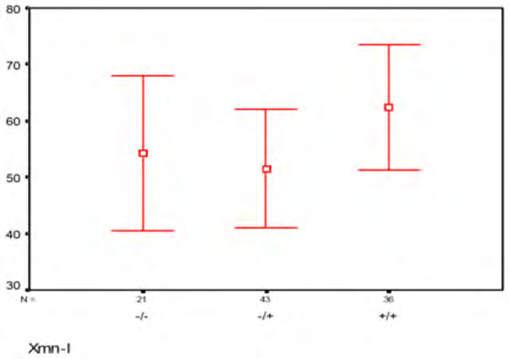
Results
Strong association of XmnI polymorphism and HbF was found. Patients being homozygous or heterozygous for the mutation had more than 80% fetal hemoglobin. This suggests that -158 GγXmnI polymorphic site results in the stimulation of fetal hemoglobin (Table 1).
| HbF % | Xmn-I -/+ genotype (%) | Xmn-I +/+ genotype (%) | Xmn-I -/- genotype (%) |
|---|---|---|---|
| 0 - 5 | 1 | 0 | 0 |
| Jun-20 | 7 | 5 | 5 |
| 21 -40 | 10 | 9 | 13 |
| 41 - 60 | 9 | 3 | 6 |
| 61 - 80 | 3 | 5 | 6 |
| 81 - 100 | 12 | 15 | 10 |
Table 1: levels of hemoglobin F in Xmn-I genotype.
Discussion & Conclusion
High hemoglobin F was found in the thalassemia intermedia patients included in this study. Milder phenotype of the disease has been associated to increased hemoglobin F. XmnI polymorphism (C-T) is associated with Gγ-globin gene affiliated with higher production of HbF [6]. XmnI polymorphism is one of the factors ameliorating β-thalassemia phenotype by stimulating fetal hemoglobin expression [7, 8].
Transcriptional silencing of γ genes was impaired in patients being heterozygous or homozygous for beta chain mutations [9]. When β-mRNA is very much decreased γmRNA and γ gene expression was found to be increased. Genetic determinant of high HbF are linked to intergenic haplotype T and does not disrupt intergenic transcription. Thus the polymorphic microsatellites (AT)x, (T)y -530 bp 5’ to β-globin gene cap site has been reported to play a role in the HbF increase, when associated with a positive XmnI site in Gγ-globin gene promoter [9]. In Thalassemia intermedia hemoglobin F percentage seems to play an important role [10]. In thalassemia reduced or absence of beta globin chains results in the formation of its clinical grades which are β-thalassemia major and beta-thalassemia intermedia. Reduced beta globin chains results in reduced production of functional HbA. This leads to ineffective erythropoiesis causing anemia. Anemia stimulates erythropoietin secretion and produce erythroid hyperplasia.
This proceed to the production of red cells precursors with fetal globin gene and formation of HbF in the postnatal life. Increased HbF in thalassemia causes increased production of F-cells. In this condition continuous production of HbF is essential for the balance of alpha/non-alpha globin ratio which reduces bone marrow hyperplasia and ineffective erythropoiesis and therefore lowers the severity of disease [4].
HbF in adults in indiscriminately distributed. The cells with increased α chains will have increased γ chain synthesis to balance the globin chains so as to survive in the bone marrow and to be released in blood circulation. This factor along with erythroid expansion likely accounts for increased fetal hemoglobin, however other factors may also be involved in the alteration of HbF production [11].
Level of hemoglobin F is affected by many loci inside or outside the β-globin gene cluster. BCL11A and HBS1L-MYB loci have a minor effect on HbF level compared to the XmnI. Homozygous or compound heterozygous states for mild alleles have much higher Hb F levels than those heterozygous states for β-thalassemia [12].
Incomplete switch over of fetal to adult hemoglobin synthesis which leads to continued synthesis of HbF throughout adult life is also one of the reasons of high hemoglobin F. This residual γ-globin expression in adults is found in some of the red cells called F cells (FC). The levels of HbF and FC are found to be variable in different population. About 13- 32% of FC are increased if associated to XmnI-Gγ site. Chromosome 6q23 and chromosome Xp22.2-p22.3 are also found to be associated to adult Hb F and FC levels [13].
A network of transcription factors and their coactivators which function within multiprotein complexes are involved in the increase of γ-globin expression. Thus expression of Gγ-globin gene can be fine-tuned by XmnI-Gγ site by FC trait and has also been found to be associated to a locus on chromosome 8q and the relationship depends upon the effects associated with the XmnI-Gγ site [13].
Acknowledgement
I would like to extend my deepest gratitude to Dr Moinuddin of Baqai medical university and Gen ® Suhaib Ahmed of Arms forces institute of Pathology for their supervision and thanks are also due for the technical staff of Arms forces institute of Pathology.
References
-
Amel HK, Madeleine M, Sandrine L, Abderrahim K, Pascale P, et al. (2011) Xmn I polymorphism associated with concomitant activation of Gγ and Aγ globin gene transcription on a β0-thalassemia chromosome. Blood Cells Mol Dis 46(2):133-138.
-
Fadwa S, Amina AS (2015) XmnI polymorphism: Relation to β-thalassemia phenotype and genotype in Egyptian Children. Egyptian Journal of Medical Human Genetics 16(2): 123-127.
-
Isabela SC, Gisele C, Claudia RB (2011) XmnI polymorphism frequency in heterozygote beta thalassemia subjects and its relation to Fetal hemoglobin levels. Rev Bras Hematol Hemoter 33(6): 483.
-
Fabrizio M, Guido M, Maria PC, Enrica F, Carmelo D, et al. (2002) Factors regulating Hb F synthesis in thalassemic diseases. BMC Blood Disorders 2: 2.
-
Thein SL, Hesketh C, Wallace RB, Weatherall DJ (1988) The molecular basis of thalassemia major and thalassemia intermedia in Asian Indians: application to prenatal diagnosis Br J Haematol 70(2): 225-231.
-
Fatima ZA, Achraf L, Naima GN, Amina B, Mohcine BM (2020) XmnI Polymorphism in Sickle Cell Disease in North Morocco. Hemoglobin 44(3): 190-194.
-
Miri Moghaddam E, Bahrami S, Naderi M, Bazi A, Karimipoor M (2017) Xmn1-158 γGVariant in B-Thalassemia Intermediate Patients in South-East of Iran. Int J Hematol Oncol Stem Cell Res 11(2): 164-171.
-
Ravindra K, Anupriya K, Sarita A (2014) Influence of Xmn 1Gγ (HBG2 c.-211 C→T) Globin Gene Polymorphism on Phenotype of Thalassemia Patients of North India. Indian J Hematol Blood Transfus 30(4): 286-290.
-
Papachatzopoulou A, Kourakli A, Makropoulou P, Kakagianne T, Sgourou A, et al. (2016) Genotypic heterogeneity and correlation to intergenic haplotype within high HbF beta-thalassemia intermedia. Eur J Haematol 76(4): 322-330.
-
Aessopos A, Kati M, Tsironi M, Polonifi E, Farmakis D (2006) Exchange blood transfusions for the treatment of leg ulcerations in Thalassemia intermedia. Haematologica 91(5): ECR11.
-
Anuja P, Shanthimala D, Mahinda A, Nancy O, Laura M, et al. (2004) Thalassemia in Sri Lanka: a progress report. Human Molecular Genetics 13(2): 203-206.
-
ThiKhanh TN, Philippe J, Claire B, Mustapha M, Nathalie B, et al. (2010) The XmnI Gγ polymorphism influences hemoglobin F synthesis contrary to BCL11A and HBS1L- MYB SNPs in a cohort of 57 β-thalassemia intermedia patients. Blood Cells Mol Dis 45(2): 124-127.
-
Chad PG, Thanusak T, Steve B, Lisa C, Swee LT (2002) Evidence of Genetic Interaction between the β-Globin Complex and Chromosome 8q in the Expression of Fetal Hemoglobin. Am J Hum Genet 70(3): 793-799.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies