Takayasu Arteritis in a Female- A Rare Case Report from Rural India
Takayasu arteritis is a rare large vessel vasculitis of autoimmune origin usually affecting young women. Here we describe a young female who presented primarily for dyspnea, polymyalgia, tingling sensation of limbs and headache since 1 year. She was diagnosed with Takayasu arteritis. Her evaluation revealed pulseless radial, brachial arteries and hypertension Computed tomography based aortic angiography revealed critical stenosis and surrounding inflammation in the branches of aorta. This case highlights how the diagnosis may be delayed due to nonspecific symptoms and how symptoms of early and late stage may coexist in the patient. Early diagnosis requires high degree of suspicion, delay in diagnosis can lead to critical stenosis and obstruction of aorta and it’s branches requiring surgical intervention.
Case Summary
A 37 year old female was brought to us with symptoms of easy fatiguibility, tingling in all 4 limbs, dyspnea on exertion NYHA grade 2, headache and dizziness on and off since 2 years.
The patient had been treated at a private nursing home a year back for similar complains and was diagnosed to have severe anemia for which in the last 1 year she received 3 units of packed RBC infusion, exact etiology of anemia was not investigated but the patient did give h/o menorrhagia. Presently she was on few multivitamins and hematinics.
She denied prolong fever, loss of weight, angina or syncope. Her physical examination revealed her to be conscious, alert, pale but when we tried to take BP we were unable to obtain it by palpatory or auscultatory method in both arms, we noticed that both her radial and brachial arteries are not palpable, the extremities were not cold and there was no evidence of gangrene, the patient on lead questioning gave history of intolerance of hands to cold weather but no h/o classical raynauds phenomena. Her BP recorded in both lower limbs was right popliteal-170/90 mmhg, left popliteal-164/90 mmhg. Pulses of lower extremity were well felt bilaterally.
There was also a bruit palpated over right carotid artery.
Rest of the systemic examination was within normal limits.
Her blood investigations revealed moderate anemia, microcytic, hypochromic with raised ESR (110) and CRP (59), her ECG showed normal sinus rhythm, her trans thoracic echocardiography revealed normal size and functioning heart chambers.
In v/o pulseless arteries and a palpable bruit, CT angiography was done which showed “diffuse circumferential thickening with abnormal wall enhancement and adjacent fat inflammation noted in the right brachicephalic trunk and 5 to 6 cm segment of the proximal part of the right subclavian artery, common carotid artery causing severe luminal narrowing upto 80 to 85%. Similar findings were also observed in left subclavian artery, common carotid artery and involvement of left vertebral artery causing severe luminal narrowing.” Thoracic, abdominal aorta and renal arteries were normal in structure and calibre (Figures 1- 3).
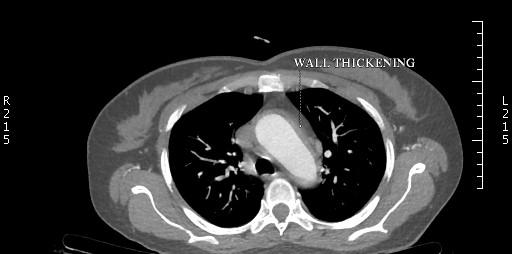
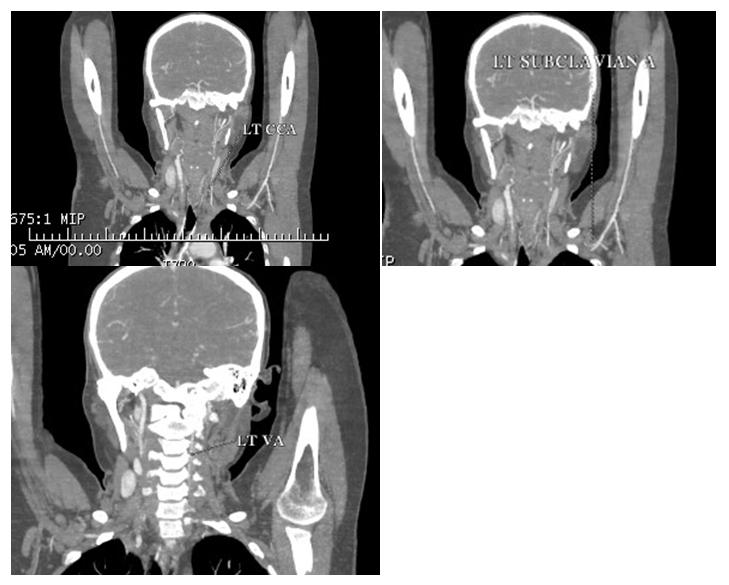
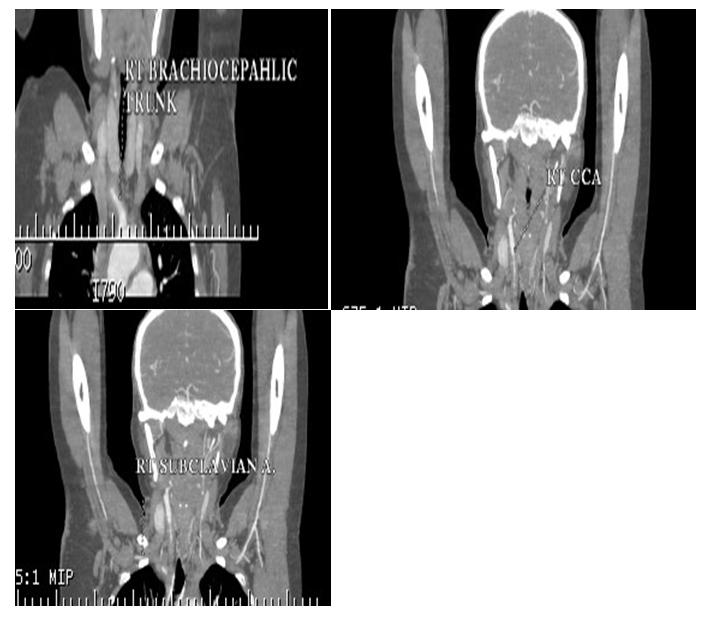
The patient was suspected to have Type 1TA. As per the criteria of American college of rheumatology patient satisfied 4 criterias classifying her for TA. The patient had no e/o active pulmonary tuberculosis and HIV and hepatitis b was ruled out following which she was started on tablet prednisolone at 1 mg/kg dose along with low dose weekly methotrexate at 5 mg weekly.
For control of hypertension she was started on tab diltiazem 30 mg TDS and tab benidipine 4 mg BD.
After 3 days of observation the patient was discharged with reduction in her symptoms of fever, myalgia and fatigue and a BP of 140/90 mmhg recorded in lower limbs.
On her follow up after 7 days the patient reported improvement in her symptoms of dizziness, blurring of vision, headache and some improvement in her headaches. Her follow up Hemoglobin had improved and inflammatory markers showed reduction (ESR 30 and CRP 17).
We plan to refer her to a vascular surgeon for opinion regarding the feasibility of surgical revascularization and its optimal timing.
Discussion
Takayasu arteritis is a rare, systemic, inflammatory large-vessel vasculitis of unknown etiology that most commonly affects women of childbearing age. It is defined as "granulomatous inflammation of the aorta and its major branches" by the Chapel Hill Consensus Conference on the Nomenclature of Systemic vasculitis [1].
Takayasu arteritis is believed to be a rare disease of young females of reproductive age group, with its incidence varying from 1 to 2 per million in Japan to 2 per million in Kuwait and 0.4 to 1.5 per million in European region, it has a worldwide distribution but is more common among females of Asian origin. In recent years there has been increased reporting from the European region also. The prevalence reported is highest in Japan that is 40/million [1].
The exact etiological and predisposing factor for TA is still to be identified. It is largely believed to be an autoimmune response primarily cell mediated influenced by environmental and genetic factors. Also an autoimmune basis is suspected because it tends to occur in various autoimmune disorders like systemic lupus erythematosis, rheumatoid arthritis, wegners granulomatosis, inflammatory bowel disease etc [2] also a possible relation between myocobacterium tuberculosis and TA has been proposed though not well substantiated [3].
It is hypothesised that in response to an unknown stimulus, aortic tissue expresses heat shock protein HSP- 65, which in turn induces Major Histocompatibility Class I Chain-Related A (MICA) on vascular cells. As a result of this γδ, αβ T cells and NK ( natural killer) T cells get activated and target this molecule by releasing perforin which initiates vascular inflammation, further release of proinflammatory cytokines leads to recruitment of mononuclear cells and further augmentation of inflammation and vascular damage. This finally leads to acute inflammation, necroses, neo-vascularization, smooth muscle migration, intimal proliferation, and giant cell formation. It is also suspected that dendritic cells along with B lymphocytes may induce production of anti- endothelial, anti-cardiolipin, and anti-aorta auto- antibodies, which accentuates the inflammatory process [4]. Classically TA progresses through three stages Stage 1: Marked by non specific constitutnal symptoms like fever, headache, myalgia, fatigue, night sweats and loss of weight. Stage 2: Vasculitic stage marked by symptoms of tenderness over the affected artery, dyspnea, palpitations, headaches, hemoptysis. Other symptoms may include arm numbness, claudication, double vision, syncope, stroke, transient ischemic attacks, hemiplegia, and paraplegia. Stage 3: Fibrotic, sclerotic or burnt out phase Characterized by remission of inflammation, symptoms due to arterial occlusions or stenosis may appear at this stage (so called pulseless disease) [5].
As the initial symptoms of the disease are nonspecific it requires high degree of suspicion and is often diagnosed late.
Specific diagnostic criteria has been described by Ishikawa (Figure 4) [2] and America college of Rheumatology (Figure 5) [6].


There are no specific blood tests to diagnose TA, ESR and CRP may be elevated earlier in the course of disease but are nonspecific. While autoantibodies like RA, ANA, ANCA are as common as in general population while antibodies like antiaorta are seldom tested. Research suggests that VCAM-1 levels are significantly elevated in cases of TA specially in elderly compared to young cases thereby suggesting that in future it may serve as a biological marker for disease activity and help in early diagnosis of TA as age progresses. Other commonly observed non specific findings are hypoalbuminemia, increase globulin and fibrinogen levels [7].
Various modalities of imaging are now available to image aorta and its branches in TA including Colour Doppler and contrast enhanced ultrasound, conventional angiography, CT angiography and MR angiography.
Conventional angiography has been traditionally considered as the gold standard for diagnosis, However, multidetector CT angiography (CTA) has emerged as a reliable tool in non-invasive assessment of both luminal and mural lesions in the aorta and its main branches, which may facilitate the detection of vasculitis during the early phase of TA [7]. Based on angiographic findings TA is classified into 5 types (Figure 6) [8]. • Type I involves only the branches of the aortic arch.
- Type IIa involves ascending aorta, aortic arch and its branches.
- Type IIb affects ascending aorta, aortic arch and its branches, and thoracic descending aorta.
- Type III involves the descending thoracic aorta, the abdominal aorta and/or the renal arteries. The ascending aorta, the aortic arch and its branches are not affected.
- Type IV involves only the abdominal aorta and/or renal arteries.
- Type V has combined features of Type IIb and IV.
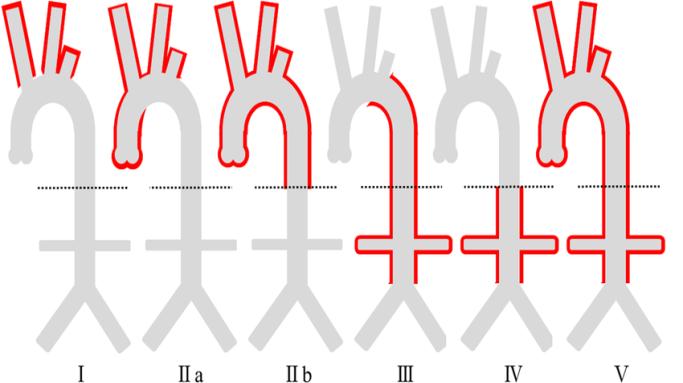
Of these type V is most commonly seen. Management can be medical or surgical. 2 main aims of medical therapy is to control inflammatory process and hypertension.
Corticosteroids form the mainstay of medical therapy which need to be given for prolong period, some patients who become either steroid dependent or are do not respond well to steroids are given cytotoxic drugs like methotrexate, azathioprine, cyclosporine.
Recently tocilizumab a humanized monoclonal antibody been proposed as a treatment for Takayasu arteritis through blockade of the soluble interleukin-6 (IL-6) receptor as IL-6 is a major component in the proinflammatory process of large-vessel vasculitis.
Critical stenotic lesions require surgical treatment either by angioplasty or surgical revascularisation.
Indications for surgical treatment are
- Renovascular stenosis causing hypertension
- Coronary artery stenosis leading to myocardial ischemia
- Extremity claudication induced by routine activity
- Cerebral ischemia and/or critical stenosis of 3 or more cerebral vessels
- Aortic regurgitation
- Thoracic or abdominal aneurysms larger than 5 cm in diameter
- Severe coarctation of the aorta
References
-
Onen F, Akkoc N (2017) Epidemiology of Takayasu arteritis. Presse Med 46(7-8 Pt 2): e197-e203.
-
Vaideeswar P, Deshpande JR (2013) Pathology of Takayasu arteritis: A brief review. Ann Pediatr Cardiol 6(1): 52-58.
-
Roberts JR, Monteagudo LA, Shah PA, Stitt R (2018) Takayasu Arteritis: Practice Essentials, Background, Pathophysiology.
-
Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z (2011) Pathogenesis of Takayasu's arteritis: a 2011 update. Autoimmun Rev 11(1): 61-67.
-
Manfrini O, Bugiardini R (2006) Takayasu's arteritis: A case report and a brief review of the literature. Heart Int 2(1): 66.
-
Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, et al. (1990) The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 33(8): 1129-1134.
-
Roberts JR, Monteagudo LA, Shah PA, Stitt R (2018) Takayasu Arteritis Workup: Approach Considerations, Diagnostic Criteria, Assessing Disease Activity.
-
Zhu FP, Luo S, Wang ZJ, Jin ZY, Zhang LJ, et al. (2012) Takayasu arteritis: imaging spectrum at multidetector CT angiography. Br J Radiol 85(1020): e1282-e1292.
- Anomalous Origin of the Left Coronary System from the Right Coronary Cusp: A Rare Coronary Anomaly in a Patient Undergoing Aortic Stenosis Workup
- Optimizing the Therapeutic Potential of Sacubitril/Valsartan: The Promise of Co-Crystal Engineering
- Association between Mortality and Topography of Peripheral Arterial Disease due to Atherosclerosis Obliterans
- Mitral Valve Replacement vs Mitral Valve Repair
- Characteristics and Evolution of Patients with Heart Failure Hospitalized in the Cardiology Department of Dalal Jamm Hospital
- Distribution of Association between Basic Knowledge of Chest Pain and Myocardial Infarction (Heart Attack) and Demographic Variables: A Survey-Based Study