Revelation of the Involvement of Rv1651c of Mycobacterium tuberculosis H37Rv in Carbohydrate Metabolism
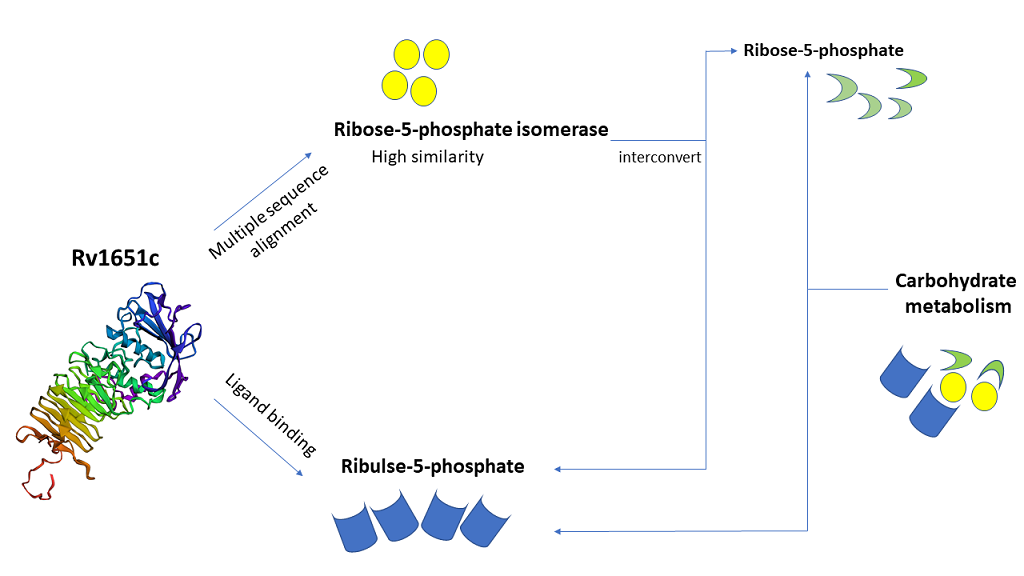
Tuberculosis (TB) is the major cause of mortality across the world. About one-third of world population is affected by this fatal disease. Mycobacterium tuberculosis H37Rv (M. tuberculosis) which is a gram- positive bacterium is responsible for the cause of TB.M. tuberculosis is spreading its roots worldwide with the help of various survival mechanisms and making its cure more difficult. In the present study, we have made use of various in-silico tools to predict the properties of Rv1651c which is a member of the PE_PGRS protein family. This manuscript reveals some important aspects of Rv1651c as its function is still unknown. The major part of this study includes protein sequence retrieval, multiple sequence alignment, protein-protein interaction study, epitope prediction, localization, function prediction, structure prediction and its validation, ligand binding prediction and mutational analysis. This protein shows the presence of GTP-binding motifs such as DXXG and GXXXXGK. These motifs can be targeted to mutate the protein and thereby, decrease its stability. This protein also shows similarity with enzyme ribose-5-phosphate isomerase, which performs the function of interconversion of ribose-5-phosphate and ribulose-5-phosphate. This similarity proves to be of great importance as this protein has ribulose-5-phosphate as one of its predicted ligands. All these in-silico generated results of Rv1651c give a hint of it being involved in carbohydrate metabolism. Carbohydrate metabolism is an important process required for the production of energy molecules. Thus, this protein might be targeted to block the carbohydrate metabolism pathway. These prediction-based studies using computational approach might prove to be a successful step towards developing drugs against TB.
Introduction
One-third of the world population is affected with tuberculosis (TB) which is a fatal disease. TB is caused by aerobic bacterium Mycobacterium tuberculosis H37Rv (M. tuberculosis) [1]. According to World Health Organization (WHO) report 2018, HIV-TB co-infection evolved as a global threat as it caused death of 1.3 million HIV negative people and 300000 HIV positive people. 27% of the Indian inhabitants are infected with TB. Antibiotic resistance is also a major havoc as 558000 people infected with TB developed resistance to rifampicin which is considered to be most effective 1st line drug of TB. Out of the rifampicin resistance cases, 82% were with multi drug resistant-TB (MDR-TB) [2]. While MDR-TB first emerged in the early 1990s, in India, the first case of extensively drug-resistant (XDR-TB) was described in 2006. Focus is now more on totally drug resistant- TB (TDR-TB). The first two cases of TDR-TB were reported by Migliori in Italy in 2007 [3]. The bacterium is breath in by healthy person and enters the lungs where it causes primary infection. It persists in alveolar macrophage for a long time and lead to the cause of active TB [4]. Sometimes, it forms a granuloma like structure after replication and remains in latent phase by hiding itself inside the granuloma [5]. In more than half of the patients, latent infection is more common and usually reaches a stage, which when left untreated leads to death [6]. The world’s most widely used vaccine against TB is Bacille Calmette–Guérin (BCG) over the past 50 years [7]. The distinct lipid cell wall of this acid-fast bacillus makes it highly unique. Its cell wall is mainly composed of glycolipids and mycolic acids but lack phospholipids containing outer membrane and therefore, does not retain dye on staining. M. tuberculosis cell wall also contains peptidoglycan, phosphatidylinositol mannosides (PIM), lipoarabinomannan (LAM), phthiocerol dimycocerosate, sulfolipids, cord factor and wax D [8, 9, 10, 11]. Genome sequencing of M. tuberculosis made understanding easy for Pro-Glu (PE) and Pro-Pro- Glu (PPE) family. The PE and PPE motifs are conserved. PE family constitutes proline and glutamic acid at 8 and 9 positions respectively. The PPE and PE family has major polymorphic tandem repeats (PPEMPTR) and polymorphic GC-rich repetitive sequence (PE_PGRS) as subsets. There are 68 members in PPE family having N-terminal and C-terminal in which N-terminal is conserved and has domain of 180 amino acid residues whereas C-terminal is variable. The PE sub family has 37 members and PE_PGRS has 63 members. PE_PGRS group contain polymorphic GC rich domain at C-terminal whereas N- terminal is conserved [12, 13, 14, 15]. Precise role of PE_PGRS protein is not clear however they play vital role in the immune-pathogenesis as they are cell surface molecules presenting antigenic variation. In PE_PGRS of M. tuberculosis calcium binding and fibronectin-binding property is also witnessed [16]. PE_PGRS protein family has GGXGXD/NXUX which is predicted to be a calcium binding site [17]. Ca2+ plays an important role in cell signaling and other significant cell processes. The fusion of phagosome and lysosome in the neutrophils is dependent on Ca2+ but M. tuberculosis has the ability to inhibit the rise of cytosolic Ca2+ and thus, prevent phagosome maturation [18]. GTPases also play significant role in growth and development of bacteria. GTP-binding proteins specifically bind and hydrolyze GTP, which in turn activate or inactivate GTPase in a cyclic manner [19]. GTP-binding domains consist of 3 consensus sequence GXXXXGK, DXXG and NKXD [20]. In Rv1651c, calcium binding motifs (GGXGXD/NXUX) and GTP binding motifs (GXXXXGK and DXXG) are present. We also found homology between Rv1651c and ribose-5-phosphate isomerase which showed various regions of similarity.Ribose-5-phosphate isomerase interconvert ribose-5-phosphate and ribulose-5-phosphate. This reaction allows the synthesis of ribose from other sugars, as well a means for salvage of carbohydrates after nucleotide breakdown [21]. The predicted similarity between Rv1651c and ribose-5-phosphate isomerase is of great importance since Rv1651c has ribulose-5-phosphate as one of its predicted ligands. This gives a hint of Rv1651c being involved carbohydrate metabolism. Carbohydrate metabolism is a very crucial process involved in the production of energy which is required by cell. Thus, this protein ‘Rv1651c’ might be targeted to block the carbohydrate metabolism pathway. In the present study, we have tried to explore different properties of Rv1651c using in-silico tools. This study might prove to be helpful in the development of successful therapy against this life-threatening disease.
Highlights
- The protein Rv1651c is a part of PE_PGRS protein family.
- It is involved in sugar metabolism according to in-silico analysis.
- This protein shows the presence of GTP-binding motifs along with calcium binding motifs.
- Mutational analysis shows large decrease in the stability of protein.
- Ligand binding prediction suggests ribulose-5- phosphate as one of the specific ligands which is involved in sugar metabolism.
Methodology
We have used various in-silico tools to find out the properties of this protein Rv1651c such as protein sequence retrieval, multiple sequence alignment, epitope prediction, localization, function prediction, protein modelling, ligand binding prediction and mutational analysis.
Protein Sequence Retrieval
The genome of M. tuberculosis can be obtained by using various servers such as Mycobrowser, UniProt, NCBI etc. We retrieved the physicochemical properties of Rv1651c from Uniprot which provided the FASTA format of PE_PGRS30 gene [22, 23].
Multiple Sequence Alignment
Multiple sequence alignment of our gene Rv1651c of M. tuberculosis was done with ribose-5-phosphate isomerase (rpiB) and its orthologues Mycobacterium bovis (M. bovis), Mycobacterium leprae (M. leprae), Mycobacterium marinum (M. marinum) and Mycobacterium smegmatis (M. smegmatis). We used MUSCLE server for the multiple sequence alignment of our gene. MUSCLE stands for Multiple Sequence Comparison by Log-Expectation and it has been acknowledged to be better in normal precision and preferred speed over ClustalW2 as well as T-Coffee server [24, 25].
Protein-protein Interaction
Proteins and their interaction with other molecules form the backbone of the machinery of the cell. The protein interaction network needs to be considered for the full understanding of biological phenomena. The Search Tool for the Retrieval of Interacting Genes (STRING) database aims to collect score and integrate all publicly available sources of protein-protein interaction information and complement them with computational predictions. It helps to find out the direct (physical) as well as indirect (functional) interactions [26]. Search Tool for Interacting Chemicals (STITCH) is a biological server that integrates the disparate data sources for 430000 chemicals into a single, easy-to-use resource. It also helps to find out the binding affinities of chemicals, which enables the user to find out the potential effect of chemicals on its interaction partners [27]. The values <0.4 means low interaction, between 0.4 to 0.7 means medium interaction and >0.7 means high interaction [28].
Epitope Prediction
To predict the B-cell epitopes in the antigen sequence, we used ABCpred server. This is the first server to be developed on the basis of recurrent neural network (machine-based technique) by using fixed length patterns. The users can select window length of 10, 12, 14, 16 and 20 as the predicted epitope length and the server is able to predict epitopes with 65.93% accuracy [29]. The identification and characterization of B-cell epitopes play important role in vaccine design, immunodiagnostic tests and antibody production. Therefore, we used BCpreds for B-cell epitope prediction [30]. We used HLApred to find out HLA class I and class II binders. This server allows the identification and prediction of 87 alleles, out of which 51 belong to Class I and 36 belongs to Class II which can be putative vaccine candidates [31].
Prediction of Sub- cellular Localization
It is important to analyze the localization of protein as it is helpful in providing the functional information about the protein. We used support vector machine (SVM) based server, TBpred to predict the sub-cellular localization of our protein. The server helps in determining if the protein is integral membrane protein, cytoplasm protein, lipid anchored protein or secretory protein by exploiting various characteristics like dipeptide composition, amino acid composition and position specific scoring matrix (PSSM). Accuracy of this SVM based server is 86.62% [32]. To further analyze the properties and sub cellular localization of our protein, we used CELLO2GO server. It is publicly accessible web-based server. The server works by combining two approaches i.e. BLAST homology and CELLO localization. CELLO2GO search for homologous sequence by making use of BLAST. This server provides result in the form of pie chart by combing molecular function, biological process and cellular compartment. By making use of these two servers, our protein was found to be present in the cell wall [33].
Signal Peptide Cleavage Site Prediction
Proteins which are synthesized newly have short sequence of amino acid at the amino terminals which are known as signal peptides (SPs). These signal peptides target the protein into or across the membrane. Signal peptide presence and their cleavage site in protein can be predicted by using SignalP4.0 server from Gram-positive bacteria, Gram-negative bacteria, archea and eukarya. It is a simple online tool which requires FASTA format. In the SignalP4.0 server, FASTA format of the protein sequence is required and other criteria required to fulfill successful running of the program include organism group, D-cutoff value, graphical output, output format, method and positional limit [34].
Graphical Abstract
Phosphorylation Site Prediction
Cellular signaling process is affected by protein phosphorylation at threonine, tyrosine and serine sites. DEPP (Disorder Enhanced Phosphorylation prediction) which is an SVM based server predicts phosphorylation at threonine, tyrosine and serine sites with sensitivity ranging from 69% to 96% [35, 36].
Transmembrane Topology and Localization Prediction
Transmembrane proteins topology and localization of helical transmembrane can be predicted by using HMMTOP. It helps in enhancing the prediction accuracy by enabling us to enter additional information regarding segment localization. HMMTOP works on the principle that transmembrane proteins topology is resolute by utmost deviation of amino acid composition of the sequence segment [37].
Function Prediction
To predict the function of our protein Rv1651c, we used VICMpred server. VICMpred is a webserver that aids in functional classification of proteins of bacteria into various divisions such as virulence factors, information molecule, cellular process and metabolism molecule. The VICMpred server makes use of SVM based method having patterns, amino acid and dipeptide composition of bacterial protein sequences. The overall accuracy of this server is 70.75% [38].
Protein Structure Prediction and Validation
We used phyre2 server for the modeling of our protein. Phyre2 is a collection of tools which are available on the web to predict and analyze protein structure, function and mutations. The focus of Phyre2 is to provide a simple and intuitive interface to state-of-the-art protein bioinformatics tools. Phyre2 makes use of advanced remote homology detection methods to build 3D models for the provided protein sequence [39]. After protein modeling, we used SAVES server to validate our protein model. SAVES server runs 6 programs to validate the protein model. It includes ERRAT, Verify3D, prove, PROCHECK, WHATCHECK and CRYST. We used ERRAT, Verify3D and RAMPAGE to validate our protein model. Ramachandran plot analysis is done using RAMPAGE server. It shows the position of residues in allowed region, disallowed region, favored region and other regions. All these programs were used to select the finest model of the protein [40].
Ligand Binding Prediction
Protein-ligand binding site identification is crucial to protein function annotation and discovery of drugs. To find out the ligands that bind with our protein, we used COACH server. COACH makes use of two new developed methods, one of which is based on binding-specific substructure comparison (TM-SITE) and another based on sequence profile alignment (S-SITE), for complementary binding site predictions. These methods have been tested on a set of 500 non-redundant proteins harbouring 814 natural, drug-like and metal ion molecules. This method successfully finds out more than 51% of binding molecules with average Matthews correlation coefficient (MCC) higher than other methods such as COFACTOR, FINDSITE and ConCavity. COACH is a consensus approach which combines TM-SITE and S-SITE with other structure-based programs and increases the MCC by 15% over the best individual predictions [41]. BioLip is a semi-manually curated database used to find out biologically relevant ligand–protein interactions. Most of the ligand binding prediction servers use the protein structures from Protein Data Bank (PDB) as templates [42]. We also used ProBis to find out the ligands that bind to Rv1651c. ProBiS-CHARMMing is a server that connects ProBiS and CHARMMing web servers into one functional unit and enables the prediction of protein-ligand complexes. This allows the optimization of geometry and the energy calculation of protein-ligand interaction. The ProBiS server predicts ligands like small compounds, proteins, and nucleic acids etc. that bind to our query protein. This is done by comparing the surface structure against a database of protein structure and finding those proteins which have similar binding sites like that of our query protein [43].
Mutational Analysis
To predict the effect of mutation on our protein, we used EASE-MM server. Evolutionary, Amino acid, and Structural Encodings with Multiple Models (EASE-MM) comprises five specialised support vector machine (SVM) models and make the final prediction from a consensus of two selected models which are based on the predicted secondary structure and accessible surface area of the mutated residue. Protein engineering and characterization of non-synonymous single nucleotide variants (SNVs) require accurate prediction of protein stability changes (ΔΔGu) caused due to single amino acid substitutions [44].
Results
Protein Sequence Retrieval
The protein sequence of Rv1651c in FASTA format was derived from UniProt. The protein length of Rv1651c is 1011 amino acids and its molecular mass is 88455.1 Da. It belongs to PE_PGRS protein family, its function is still unknown but it is thought to be involved in virulence.
Multiple Sequence Alignment
We used MUSCLE server to find out the homology of our protein with ribose-5-phosphate isomerase (rpiB) which is considered as a major enzyme involved in carbohydrate metabolism. Its function is to interconvert ribose-5-phosphate and ribulose-5-phosphate. Ribose-5-phosphate isomerase (rpiB) shows a lot of variation in the sequence pattern in different organisms. However, the reason for such variation is not known. In M. tuberculosis, the motif encompassing residues 69-75 of the RpiB sequence (GGSGNGE) provides the most distinctive signature [21]. On checking the homology of our protein with rpiB and its orthologues like M. bovis, M. leprae, M. marinum and M. smegmatis, we found several regions of similarity which hints towards the involvement of this protein in carbohydrate metabolism. The most significant region of high similarity was found between residues 88-94 of RpiB and residues 862-868 of Rv1651c. We observed only slight variation in the amino acid composition which is trivial because the substitution was observed between the amino acids of similar properties. For example, leucine at position 88 of RpiB is replaced by isoleucine at position 862 of M. tuberculosis. It is an example of conservative replacement because both these amino acids are hydrophobic in nature [45]. Similarly, other replacements observed are also conservative in nature. According to National Center for Biotechnology Information (NCBI), sequences whose identity is as low as 8% may be orthologous proteins and perform the same function. There have been various cases where enzymes that perform the same function have a region of high similarity which is flanked by dissimilar regions, this accounts for the probability of RpiB and Rv1651c having similar activity [46](Figure 1).

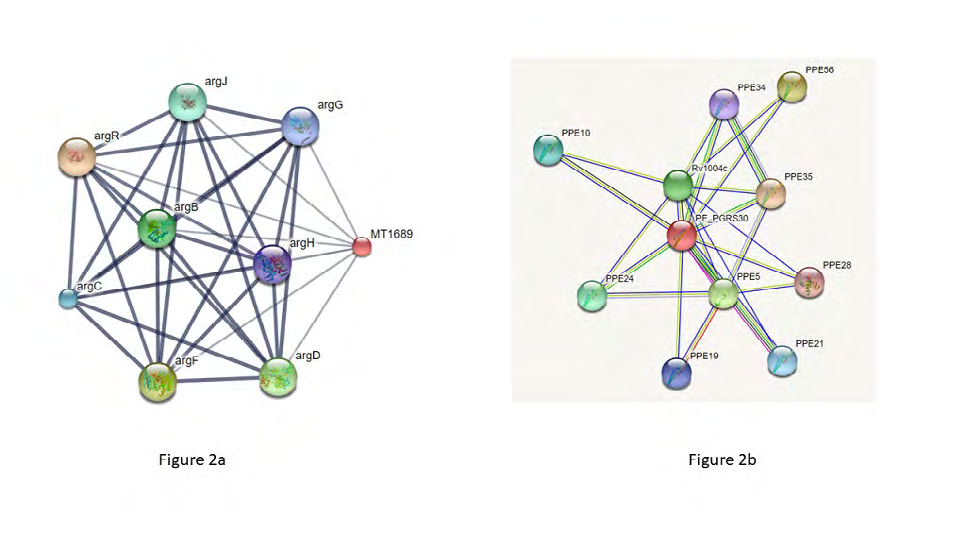
Protein-protein Interaction
STRING server shows the interaction of protein Rv1651c with mostly PPE family members such as PPE35, PPE56, PPE5, PPE24 etc. The maximum interaction is observed with PPE35 with a score of 0.921 Figure 2a. STITCH server shows interaction of protein Rv1651c with argR, argF, argD, argB, argJ, argC, argG and argH. The maximum interaction score of 0.564 is observed in the case of argR which is involved in arginine biosynthesis Figure 2b.
Epitope Prediction
To find out the epitopes present on our protein Rv1651c, we used ABCpred, BCpreds and HLApred. In ABCpred server, we selected epitope length of 16 and threshold value was selected as 0.51.Various B-cell epitopes were predicted and rank 1 was attained by sequence GGTGGSAIAFGNGGQG at start position 580 and score 0.95. BCpreds also predicted several B-cell epitopes with epitope length specified as 20 and specificity 75%. For example, epitope GAGGAGGAGSPAGAPGNGGT with length 20 and score 1 was predicted using BCpreds. The top 5 results of ABCpred and BCpreds are shown in the table (Table 1). HLApred was used to find out the MHC-II binding sites present on our protein. Various MHC-II binding sites were found by setting the threshold value 2 and number of alleles in query 9 such as HLA-A2, HLA-A*0201, HLA-A*0202 etc. Few of the HLApred results of different are shown in the figure 3.
| ABCpred | BCpreds | ||||
|---|---|---|---|---|---|
| Position | Epitope | Score | Position | Epitope | Score |
| 580 | GGTGGSAIAFGNGGQG | 0.95 | 670 | GAGGAGGAGSPAGAPGNGGT | 1 |
| 645 | TKAGGTGSDGGHGGNA | 0.94 | 164 | GGAGGAGGAGGAGGAGGAGG | 1 |
| 205 | GGAGGNALLFGNGGNG | 0.94 | 426 | GAGGAGGAGGVGGLLYGNGG | 1 |
| 627 | FGDGGTGGTGGAGGAG | 0.93 | 365 | YGNGGAGGAGGNGGDTTVPL | 1 |
| 718 | SSVPILGPYEDLIANT | 0.92 | 546 | GAGGLIWGNGGAGGNGGNGG | 1 |
Table 1: B-cell epitope prediction. This table shows the top 5 epitopes predicted using ABCpred and BCpreds server taking epitope

Prediction of Sub- cellular Localization
The SVM based server TBpred was used to determine the localization of Rv1651c which presented scores from different class wise SVM model. This SVM model showed
| Localization | Score |
| Extra cellular | 1.63 |
| Cell wall | 2.331 |
| Membrane | 0.361 |
| Cytoplasmic | 0.678 |
Table 2: Localization of Rv1651c done using CELLO2GO server.
1.4352752 as the highest score of our protein indicating that it is an integral membrane protein. To confirm our prediction, we used another sever named CELLO2GO giving the highest score of 2.331 which belonged to cell wall (Table 2).
CELLO2GO server was used to predict the localization of Rv1651c. The maximum score was 2.331 and was attained by cell wall.
Signal Peptide Cleavage Site Prediction
Signal peptide cleavage site was done using SignalP4.0 prediction server. In our study, the organism group is Gram- positive with D-Score cut-off value of 0.450.In our protein, the highest Y-Score and C-score was at 41st residue which was 0.474 and 0.488 respectively and other cleavage sites with relatively lower S-Score was 0.833 at 27th residue.
Phosphorylation Site Prediction
The number of serine, threonine and tyrosine phosphorylated sites are predicted by DEPP server. DEPP results of serine, threonine and tyrosine phosphorylation sites show that in Rv1651c protein 39 out of 68 residues are phosphorylated at serine, 12 out of 66 residues are phosphorylated at threonine and 0 out of 10 residues are phosphorylated at tyrosine. The statistic score shows
| Function | Class |
| Cellular process | -5.4308081 |
| Information molecule | -0.87052855 |
| Metabolism | -6.258689 |
| Virulence factors | -2.0960081 |
Table 3: Function Prediction using VICM server. VICM predicted the function performed by our protein Rv1651c. The maximum value o
57.3529% phosphorylated serine, 18.1818% phosphorylated threonine and 0% phosphorylated tyrosine sites.
Transmembrane Topology and Localization Prediction
The HMMTOP server predicts both the localization and topology of helical transmembrane segments. By using HMMTOP server, it is identified that gene Rv1651c forms two transmembrane helices between 828-847 and 874- 898 amino acid sequence. This indicates the position of our protein within the membrane.
Function Prediction
On predicting the functional class of our protein using VICM, it comes out be a member of information and storage molecules with a score of -0.87052855. Information and storage function lead by other classes of functions such as cellular processes and metabolism followed by virulence factors (Table 3).
Protein Structure Prediction and Validation
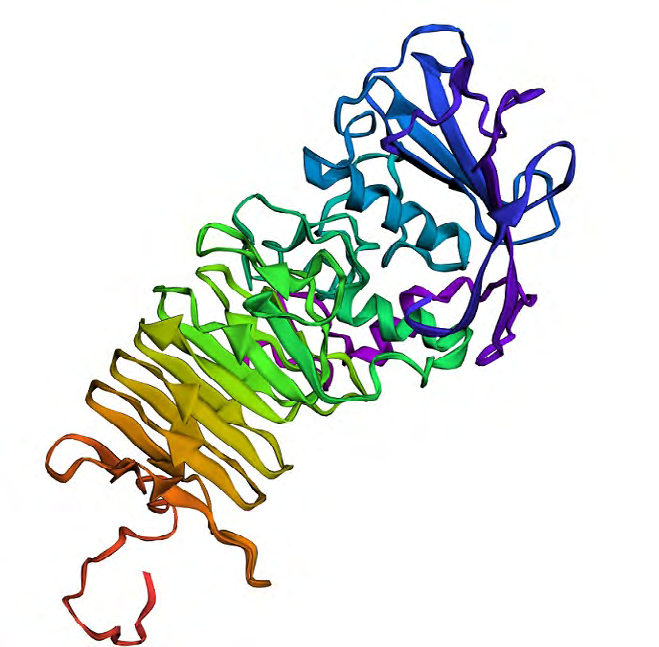
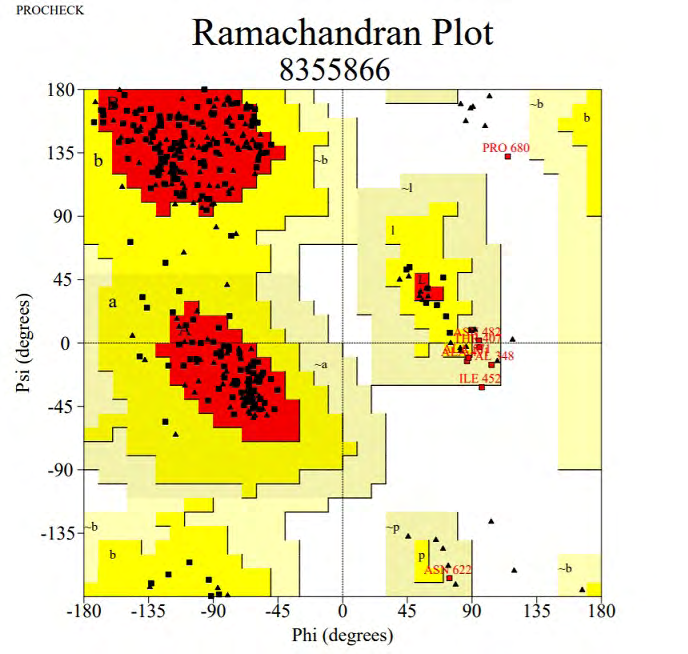
Several protein models were generated using Phyre2 server out of which we selected 5 models. The confidence score of 5 protein models was 98.9, 98.8, 98.7, 98.2 and 98.2 respectively. We selected the protein model with confidence score 98.7 which had PDB header as hydrolase, chain A, PDB Molecule as secreted protease C and PDB Title as PRTC from erwinia chrysanthemi: e189a mutant (Figure 4). This protein model was selected on the basis of validation of the 5 selected models. Validation of the protein models was done using SAVES server. We used Verify3D, ERRAT and RAMPAGE to authenticate the protein model. In Verify3D, the average 3D-1D score of more than or equal to 0.2 was observed for 99.74% of the residues. So, our protein model is passed according to Verify3D. The errat score showed the overall quality factor of protein as A: 100 B: 73.494 and C: 19.4444.RAMPAGE was used to generate the Ramachandran Plot of the modelled protein. There were 82.6% residues (A, B, L) in most favored region, 13.9% residues (a, b, l, p) in additional allowed regions, 3.0% residues (~a, ~b, ~l, ~p) in generously allowed regions and 0.5% residues in disallowed regions (Figure 5).
Ligand Binding Prediction
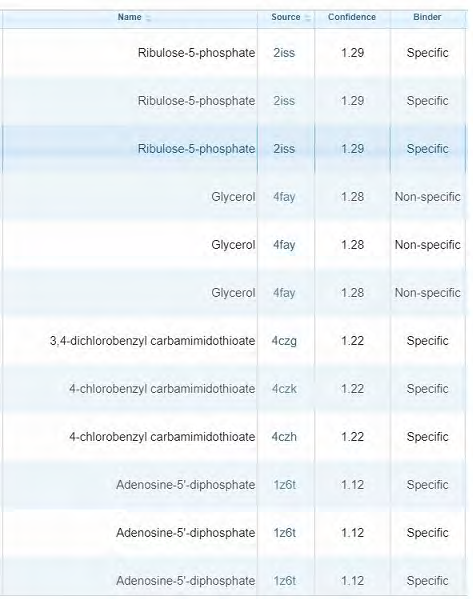
We used COACH server to find out the ligands that bind to our protein. It predicted several ligands associated with our protein. Rank 1 was attained by calcium which adds up to our prediction of the presence of calcium binding sites (motifs) in the protein. We also used ProBis server to find out the ligand binding sites. It predicted several ligands for 4 different binding sites. Ribulose-5-phosphate was one of the predicted ligands with confidence 1.29 and binding highly specific. It also adds up to our result that our protein Rv1651c shows homology with enzyme ribose-5-phosphate isomerase whose function is to interconvert ribose-5- phosphate and ribulose-5-phosphate. It also shows GTP as one of the predicted ligands with specific binding summing up our prediction of the presence of GTP-binding motifs. Various other specific and non-specific ligands are found using ProBiS. The ligands that are predicted to bind at site 1 are shown in the figure (Figure 6).
Mutational Analysis
To analyze the effect of mutation on our protein Rv1651c, we used EASE-MM server. We performed mutations at several points of the protein. The maximum destabilizing effect on the protein was observed at 76th position on the protein sequence when phenylalanine is mutated with glycine. The value of ΔΔGu observed in the case of this mutation is -5.0445 Kcal/mol. Instead of finding the effect of mutation on specific motifs, we focused on the positions where the effect of mutation was the highest. The top 5 mutations that cause highest destabilizing effect on our protein are shown in the table 4.
| Mutation | ΔΔG u (KJ/mol) | Stability class |
|---|---|---|
| F76G | -5.0445 | destabilising |
| F55G | -4.8346 | destabilising |
| Y755G | -4.7834 | destabilising |
| F62G | -4.7543 | destabilising |
| F751G | -4.6928 | destabilising |
Table 4: Mutational analysis of the protein. Mutational analysis of Rv1651c was performed by EASE-MM server. The top 5 mutations
Discussion
In spite of several treatments available, TB which is caused by M. tuberculosis still remains a global burden. There is an urgent need to combat the outcome of this hazardous disease_. M. _tuberculosis employs an uncommon methodology to reside inside its host and overcome its immunological response [1]. In our study, we predicted the properties of Rv1651c protein, which belongs to PE_PGRS protein family. This protein shows high regions of similarity with enzyme ribose-5-phosphate isomerase, which interconverts ribose- 5-phosphate and ribulose-5-phosphate [24, 25]. This similarity proves to be of great importance in our prediction study as our protein also shows ligand binding sites for ribulose-5-phosphate [43]. Epitopes present on our protein were predicted using ABCpred, BCpreds and HLApred [29, 30, 31]. The protein structure was modelled and validated using Phyre2 and SAVES server respectively [39, 40]. This protein also shows GTP-binding motifs which is highly significant as GTP-binding proteins initiate to be as novel targets for the treatment of this disease [20]. Mutational analysis shows high decrease in the stability of protein and ligand binding prediction suggests some molecules that can be used for the targeted delivery of drugs [44]. Therefore, targeting this gene for disruption of its functional characteristics and interfering the predicted carbohydrate metabolism might be a good footstep towards developing effective drug against tuberculosis.
Acknowledgement
The authors acknowledge financial support from the Department of Science and Technology- SERB, Council of Scientific and Industrial Research-Institute of Genomics and Integrative Biology under the research project GAP0145 (SERB-DST Grant no: EEQ/2016/000514).
References
-
Meena LS, Rajni (2010) Survival mechanisms of pathogenic _Mycobacterium tuberculosis_ H37Rv. FEBS J 277(11): 2416-2427.
-
WHO (2018) Global Tuberculosis report.
-
Udwadia ZF (2012) MDR, XDR, TDR tuberculosis: ominous progression. Thorax 67(4): 286-288.
-
Esmail H, Barry 3rd CE, Young DB, Wilkinson RJ (2014) The ongoing challenge of latent tuberculosis. Philos Trans R Soc Lond B Biol Sci 369(1645): 20130437.
-
Pieters J (2008) _Mycobacterium tuberculosis_ and the macrophage: maintaining a balance. Cell Host Microbe 3(6): 399-407.
-
WHO (2006) Global Tuberculosis Report.
-
Andersen P, Doherty TM (2005) The success and failure of BCG-implications for a novel tuberculosis vaccine. Nat Rev Microbiol 3(8): 656-662.
-
Fratti RA, Chua J, Vergne I, Deretic V (2003) Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A 100(9): 5437-5442.
-
Brennan PJ (2003) Structure, function, and biogenesis of the cell wall of _Mycobacterium tuberculosis_. Tuberculosis (Edinb): 83(1-3): 91-97.
-
Alderwick LJ, Birch HL, Mishra AK, Eggeling L, Besra GS (2007) Structure, function and biosynthesis of the _Mycobacterium tuberculosis_ cell wall: arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem Soc Trans 35(5): 1325-1328.
-
Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, et al. (1997) Role of the major antigen of _Mycobacterium_ _tuberculosis_ in cell wall biogenesis. Science 276(5317): 1420-1422.
-
Dheenadhayalan V, Delogu G, Brennan MJ (2006) Expression of the PE_PGRS 33 protein in _Mycobacterium_ _smegmatis_ triggers necrosis in macrophages and enhanced mycobacterial survival. Microbes Infect 8(1): 262-272.
-
Delogu G, Sanguinetti M, Pusceddu C, Bua A, Brennan MJ, et al. (2006) PE_PGRS proteins are differentially expressed by _Mycobacterium tuberculosis_ in host tissues. Microbes and Infection 8(8): 2061-2067.
-
Banu S, Honoré N, Saint-Joanis B, Philpott D, Prévost MC, et al. (2002) Are the PE_PGRS proteins of _Mycobacterium_ _tuberculosis_ variable surface antigens? Mol Microbiol 44(1): 9-19.
-
Meena PR, Monu, Meena LS (2016) Fibronectin binding protein and Ca2+ play an access key role to mediate pathogenesis in _Mycobacterium_ _tuberculosis_: An overview. Biotechnol Appl Biochem 63(6): 820-826.
-
Espitia C, Laclette JP, Mondragón-Palomino M, Amador A, Campuzano J, et al. (1999) The PE-PGRS glycine-rich proteins of _Mycobacterium tuberculosis_: a new family of fibronectin-binding proteins? Microbiology 145(12): 3487-3495.
-
Beg MA, Athar F, Meena LS (2019) Significant Aspect of Rv0378 Gene of _Mycobacterium tuberculosis_ H37Rv Reveals the PE_PGRS like Properties by Computational Approaches. J Biotechnol Biomed 2: 24-39.
-
Vergne I, Chua J, Singh SB, Deretic V (2004) Cell biology of _Mycobacterium tuberculosis_ phagosome. Annu Rev Cell Dev Biol 20: 367-394.
-
Rajni, Meena LS (2010) Guanosine triphosphatases as novel therapeutic targets in tuberculosis. Int J Infect Dis 14(8): e682-e687.
-
Joshi K, Meena S, Meena LS (2020) Analysis of predicted amino acid biosynthesis in Rv3344c in Mycobacterium tuberculosis H37Rv using bioinformatics tools. Biotechnol Appl Biochem, Mar; 67(2):213-223, doi:10.1002/ bab.1834.
-
Roos AK, Andersson CE, Bergfors T, Jacobsson M, Karlen A, et al. (2004) _Mycobacterium tuberculosis_ ribose-5- phosphate isomerase has a known fold, but a novel active site. J Mol Biol 335(3): 799-809.
-
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, et al. (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32: 115-119.
-
UniProt Consortium (2015) UniProt: a hub for protein information. Nucleic Acids Res 43: 204-212.
-
Madeira F, Park YM, Lee J, Buso N, Gur T, et al. (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47(1): 636-641.
-
Li W, Cowley A, Uludag M, Gur T, McWilliam H, et al. (2015) The EMBLEBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43(1): 580-584.
-
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, et al. (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 47(D1): D607-D613.
-
Szklarczyk D, Santos A, von Mering C, Jensen LJ, Bork P, et al. (2016) STITCH 5: augmenting protein– chemical interaction networks with tissue and affinity data. Nucleic Acids Res 44(D1): D380-D384.
-
Lewis AC, Saeed R, Deane CM (2010) Predicting protein- protein interactions in the context of protein evolution. Mol Biosyst 6(1): 55-64.
-
Saha S, Raghava GPS (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1): 40-48.
-
Chen J, Liu H, Yang J, Chou KC (2007) Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 33(3): 423-428.
-
Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L (1998) Prediction of MHC class-II binding peptides using an evolutionary algorithm and artificial neural networks. Bioinformatics 14(2): 121-130.
-
Rashid M, Saha S, Raghava GPs (2007) Support Vector Machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC bioinformatics 8: 337.
-
Yu CS, Cheng CW, Su WC, Chang KC, Huang SW, et al. (2014) CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PloS One 9(6): e99368.
-
Armenteros JJA, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, et al. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37(4): 420-423.
-
Blom N, Gammeltoft S, Brunak S (1999) Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294(5): 1351-1362.
-
Blom N, Pontén TS, Gupta, R, Gammeltoft S, Brunak S (2004) Prediction of post‐translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4(6):1633-1649.
-
Tusnady GE, Simon I (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283(2): 489-506.
-
Saha S, Raghava GPS (2006) VICMpred: an SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom Proteom Bioinf 4(1): 42-47.
-
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10(6): 845-858.
-
Lovell SC, Davis IW, Arendall III WB, De Bakker PI, Word JM, et al. (2003) Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 50(3): 437-450.
-
Yang J, Roy A, Zhang Y (2013) Protein–ligand binding site recognition using complementary binding- specific substructure comparison and sequence profile alignment. Bioinformatics 29(20): 2588-2595.
-
Yang J, Roy A, Zhang Y (2013) BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res 41: 1096-1103.
-
Konc J, Miller BT, Štular T, Lešnik S, Woodcock HL, et al. (2015) ProBiS-CHARMMing: web interface for prediction and optimization of ligands in protein binding sites. ACS Publications 55(11): 2308-2314.
-
Folkman L, Stantic B, Sattar A, Zhou Y (2016) EASE- MM: Sequence-based prediction of mutation-induced stability changes with feature-based multiple models. J Mol Biol 428(6): 1394-1405.
-
Gilles AM, Marliere P, Rose T, Sarfati R, Longin R, et al. (1988) Conservative replacement of methionine by norleucine in Escherichia coli adenylate kinase. J Biol Chem 263(17): 8204-8209.
-
Koonin EV, Galperin MY (2003) Principles and methods of sequence analysis. Sequence—Evolution—Function, Springer, pp: 111-192.
- Antifungal Activity of New Acetophenone Derivatives
- Interconnected Microbiomes Human Health Within an Environmental Framework
- Silkworm-Based Vaccine Production for H5N1: A One Health Approach to Pandemic Preparedness
- Microbial Diversity and Lipolytic Activity of Bacteria and Fungi from Oil-Contaminated Sites in Makurdi Metroplois
- Antibiotic Resistance Profile of Bacteria Isolated at the Central Laboratory of the National Hospital Center of Nouakchott
- Epidemiology and Sensitivity to Antibiotics of Germs Isolated from Blood Cultures in the Laboratory of the National Hospital Center of Nouakchott-Mauritania