Efficacy Evaluation of Meloxicam 15mg Tablet through Bioequivalence Study in Indonesian Healthy Volunteers
Objective: The objective of this study is to compare the efficacy between the generic drug of Meloxicam 15mg Tablets produced by PT. Kimia Farma Tbk and its comparator (Movi-Cox® 15mg Tablet by PT. Boehringer Ingelheim Indonesia) by a study of bioequivalence in Indonesian healthy volunteers Methods: A randomized, single-dose, open-label, crossover two-way (2 treatments, 2 periods, and 2 sequences) under fasting condition were conducted in healthy Indonesian volunteers to evaluate bioequivalence between 2 formulations of Meloxicam 15mg tablet. This study required washout period of at least 7-days. The test and comparator drug were administered to subjects as a single oral dose. Blood samples were collected adequately (0–72 h) with 18 points to determine meloxicam concentrations in plasma using a validated LC-MS/MS analytical method. The number of subjects who participated and completed the study were 18 volunteer (9 male and 9 female). The pharmacokinetic parameters calculated in this study are AUC0-72, AUC0-inf, Cmax, tmax, and t½. The bioequivalence acceptance limit of 90% confidence interval (CI) with α = 5.00% is 80.00–125.00% for AUC and Cmax parameters. The estimation of Tmax and T½ in the bioequivalence study was based on a nonparametric statistical procedure on the original data using Wilcoxon Sign Test. Result: The results for AUC0-72 and Cmax were 108.07% (104.88-111.35) % and 105.24% (97.72-113.34) % respectively with intra-subject variability (%CV) was 5.15% for AUC0-t and 12.80% for Cmax. These results were within the regulatory acceptance limit 80.00-125.00%. Conclusion: Based on this result, it was concluded that the test formulation is bioequivalent to comparator formulation.
Introduction
Indonesian Food and Drug Regulatory Authority (BPOM) is obliged to provide guarantees to the public that drugs in circulation meet the requirements for efficacy, safety and quality through supervision before and after being marketed. One of the monitoring of drugs before they are marketed is the assessment of BE test results for generic drugs as proof that the generic drug has equivalent therapeutics to its comparator drug. This BE test is mandatory for drugs classified as non-steroidal anti-inflammatory drugs such as Meloxicam. The BE test must be carried out in a BE test laboratory that meets GLP/ISO 17025 and GCP standards [1].
Concerns have been raised about the possibility that the quality of some marketed generics may not fully reflect the results obtained in pre-registration bioequivalence investigations. The substitution of branded products with generic medicines is a matter of discussion, and is often regarded with scepticism by healthcare providers and patients. Current bioequivalence requirements are based on average bioequivalence measures; however, there are concerns that the use of such measures may not be appropriate in the case of drugs with high intrasubject or inter-subject variability, such as analgesic drugs. Furthermore, in some countries, the lack of open access to pre-registration study documentation is a weak point of the generics market. Generics market Indeed, prescribing physicians cannot access information on the pre-registration development of generic drugs, and they can only trust that the regulatory authorities approve certain generic formulations according to recommended procedures. The pharmacokinatick data of Meloxicam are tabulated in Table 1.
| Parameter | Ahmad et al, 2011 | Gschwend et al, 2007 | Jiménez et al, 2005 | Tangsucharit et al, 2009 |
|---|---|---|---|---|
| Countries | Pakistan | Turkey | Mexican | Thailand |
| Cmax(ng/mL) | 1023-1051 | 1064-1146 | 2559-2290 | 1027-1151 |
| tmax (h) | 3.12-3.75 | 5 | 4.36-5.12 | 4.5 |
| t1/2 (h) | 13.31 -13.69 | 18.29 - 18.94 | 19.95 -19.05 | - |
| AUC0-t(ng ·h/ml) | 28.067-28.385 | 32699-31846 | 61230-55446 | 34.024 - 35.137 |
| %CV Cmax | - | 8.51% | 15.87% | 16.32% |
| %CV AUC0-t | - | 5.72% | 16.83% | 17.10% |
Table 1: The Pharmacokinetics data of Meloxicam. In Indonesia, no studies have been conducted on the bioequivalence of meloxicam.
Table 1: The Pharmacokinetics data of Meloxicam. In Indonesia, no studies have been conducted on the bioequivalence of meloxicam. Consequently, it is very important to conduct pharmacokinetics and bioavailability studies of meloxicam in the local population. Therefore, this study was designed to determine the bioequivalence parameters of meloxicam in the local population to ensure the rational use and safety of this good medicinal material.
Pharmacology
Meloxicam is a nonsteroidal anti-inflammatory drug (NSAID) which has anti-inflammatory, analgesic and antipyretic effects. Meloxicam is a derivative of oxicam and is included in the enolic acid class of NSAIDs. Meloxicam has the chemical name 1,1-Dioxide- 4- Hydroxy-2- methyl-N- (5-methyl-2-thiazolyl)-2H-1, 2-benzothiazine- 3carboxamide [2].
Meloxicam works by inhibiting the synthesis of prostaglandins which are inflammatory mediators through inhibiting cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). Prostaglandins cause sensitization of afferent nerves and potentiate the action of bradykinin in inducing pain. The anti-inflammatory effect is mainly due to inhibition of the COX-2 isoenzyme, although COX-1 is expressed at some sites of inflammation. Meloxicam has shown the ability to reduce the erythrocyte sedimentation rate (ESR) in rheumatoid arthritis patients. Apart from that, it can also reduce C- Reactive Protein (CRP) and aquaporin-1 expression [3]. Meloxicam is indicated for treating pain and inflammation in rheumatic diseases, worsening osteoarthritis (short term), ankylosing spondylitis (chronic inflammation that can cause the gaps between the vertebrae to close) [4].
Pharmacokinetics
Oral meloxicam is absorbed in the digestive tract with an absolute bioavailability of 89% and the time required to reach maximum concentrations is around 3-5 hours. The peak concentration of the second dose of Meloxicam occurs around 12-14 hours, which shows that biliary recycling occurs. After administration of several doses, steady state (CSS) Cmax concentrations were achieved on day 5. The average elimination half-life (t½) of oral meloxicam is approximately 13-20hours. Meloxicam binds very strongly to plasma proteins, especially albumin, up to 99.4%. The protein binding fraction was not affected by drug concentration in healthy individuals, but decreased to 90% in patients with kidney disease [3, 5].
Meloxicam is almost completely metabolized extensively in the liver, by cytochrome P450 enzymes and CYP3A4 isoenzymes. Meloxicam has 4 main metabolites which are known to have pharmacological activity in vivo, where 60% of the dose through liver cytochrome enzyme oxidation is converted into 5’- carboxy meloxicam and 5’-hydroxymethyl meloxicam. Meanwhile, 2 other metabolites are formed through peroxidase activity, which respectively reach 16% and 4% of the administered dose. The remaining unchanged drug is excreted in urine (0.2%) and feces (1.6%). Approximately 6% and 13% of the dose is found in the urine in the form of 5’-hydroxymethyl and 5’-carboxy metabolites, respectively. [3].
Side Effects
Meloxicam has side effects, namely digestive disorders (stomach pain, constipation, diarrhoea, flatulence, dyspepsia, nausea and vomiting); disorders of the circulatory and lymphatic systems (anaemia, agranulocytosis, thrombocytopenia, leukopenia); heart problems (heart failure, palpitations or heart palpitations); immune system disorders (angioedema); metabolic and nutritional disorders (hyperkalaemia, dehydration); nervous system disorders (headache, vertigo); psychiatric disorders (anxiety, depression); kidney and urinary tract disorders (haematuria, albuminuria); skin and subcutaneous tissue disorders (pruritus, rash, photosensitivity, urticaria); vascular disorders (hypertension, hypotension); and increased serum transaminase levels, increased serum bilirubin, increases serum creatinine and BUN, weight gain or loss [6].
Contraindications
Meloxicam is contraindicated in patients with hypersensitivity to meloxicam, aspirin or other NSAIDs. History of active gastrointestinal bleeding, ulceration or perforation related to NSAID use. Active inflammatory bowel disease (eg ulcerative colitis), severe heart failure Perioperative pain treatment in the setting of Coronary Artery Bypass Graft (CABG) surgery Sufferers of severe liver disorders Pregnant women (3rd trimester) and breastfeeding [6]. The objective of this study is to compare the efficacy between the generic drug of Meloxicam 15mg Tablets produced by PT. Kimia Farma Tbk and its comparator (Movi- Cox® 15mg Tablet by PT. Boehringer Ingelheim Indonesia) by a study of bioequivalence in Indonesian healthy volunteers.
Study Protocol
The protocol study has reviewed and approved by the Ethics Committee of the Faculty of Medicine, University of Indonesia (Jakarta, Indonesia) and the Indonesian Food and Drug Regulatory Authority (Jakarta, Indonesia). This study was conducted in compliance with the Indonesian Guidelines for Bioequivalence Studies and following ethical principles of the Declaration of Helsinki, GCP, and GLP standard.
Study Design
A randomized, single-dose, open-label, two-way crossover design (2treatments, 2periods, and 2sequences) were conducted in this study under fasting condition [5, 7, 8, 9]. In determining the washout period or gap period between two treatment periods so that the effects of treatment in the previous period do not transfer to treatment in the next period, the minimum washout period is 5.5 x t½. Based on previous kinetic studies, the t½ range is 13-20 hours, so the minimum time required is around 71.5-110hours or 5days [3, 4]. However, in this Meloxicam study, the washout period was set at 7 days. After 7 days, the drug was given again based on randomization results in the same way as period 1[10].
Subject Screening
The number of subjects required was calculated based on variations in the main bioavailability parameters, namely Cmax and AUC0-t in plasma in previous studies, which indicate the amount of drug entering systemic blood circulation. Based on the %CV Cmax and AUC values in the reference of previous study, the highest %CV value was 17.10%. Based on BPOM provisions, drugs with this %CV require a minimum of 14 subjects in the BE test [1]. In this study, the number of subjects included was 18 people.
Subjects involved in the study must participate voluntarily and subjects are free to withdraw from the study at any time. Subjects were given a detailed explanation in advance by the study coordinator and doctor regarding the risks and benefits of this study in January 2022. Subjects who were willing to take part in this study until completion had signed informed consent.
Within seven days before the first administration of the study is conducted, the screening is carried out to assess the volunteer’s health condition, in accordance with the inclusion and exclusion criteria. The inclusion criteria of the subjects were considered eligible for participation in this study, if: 1) Signed informed consent; 2) Healthy based on clinical laboratory tests (routine haematology, liver function, kidney function, blood glucose, urinalysis, hepatitis B (HBsAg), hepatitis C (Anti-HCV) and HIV (Anti-HIV), medical history, and physical examination); 3) Male and female subjects (if female, consider the risks for women of childbearing age and perform pregnancy tests); 4) Age between 18-55 years; 5) Normal weight range according to Body Mass Index (BMI) 18-25 kg/m2); 6) Vital signs within the following ranges: systolic blood pressure 110-129 mmHg, diastolic blood pressure 70-84mmHg, normal pulse rate 60-90 bpm, oxygen saturation (SpO2) in the normal range of 95-100%, and normal respiratory rate of 12- 20/min [1].
The exclusion criteria for this study including: 1) Smoking more than 10 cigarettes per day; 2) Pregnant or breastfeeding women (Pregnancy tests was performed during screening and prior to the administration of the investigational or comparator drug); 3) History of kidney or liver disease, or history of allergy, hypersensitivity or contraindication to the investigational bioequivalence drug; 4) Clinically significant haematological abnormalities; 5) Abnormal electrocardiogram (ECG); 6) Difficulty accessing veins in the left or right arm; 7) History of significant ongoing clinically or medically significant chronic or acute illness; 8) History of drug or alcohol abuse within the past 12 months (1 year) prior to screening for this study; 9) Positive serology test results for Hepatitis B (HBsAg), Hepatitis C (anti-HCV), HIV (anti-HIV). 10) Positive rapid antigen test results for SARS-CoV-2 (if the BE study is conducted during a pandemic); 11) Have history or condition that can affect drug kinetics; 12) Use of drugs or dietary supplements no more than 7 days since the start of the study; 13) participated in previous clinical trials no more than 3 months from the start of the study; and 14) Blood donation or blood loss of more than 300ml within 3 months from the start of the study [1].
The volunteers who enrolled in the study were 18 people (9 males and 9 females). The demographic data of the subject are tabulated in Table 2.
| Parameter | MIN | MAX |
|---|---|---|
| Age (Year) | 20 | 52 |
| Body Weight (kg) | 43 | 80 |
| Body Height (m) | 150 | 179 |
| BMI (kg/m2) | 18.37 | 24.97 |
Table 2: Demographic Data of 18 subjects.
Drug Product Administration
The subjects are instructed not to take any medication for at least 1week before the start of the study. If they have to take any medication or food supplements due to an urgent situation, the medication or supplement taken and the dosage should be reported to the investigator. The subjects are not allowed to smoke, drink alcohol, fruit juice, coffee, tea, chocolate, bread, and soda water 24hours before the start of the study. The subjects must fast for 8 hours before taking the test drug or comparator drug. About 1 hour before taking the drug, the condition of the subjects is checked by a doctor to assess their health. The examination includes blood pressure, pulse rate, body temperature, oxygen saturation (SpO2) examination, and respiratory rate. The results of the examination are recorded in the CRF. Additional examinations, such as pregnancy tests, are conducted for female subjects.
Starting at 07.00 on the first day, the subjects are given the test drug or the comparator drug with 220ml of water while sitting. The test drug in this study is Meloxicam 15mg tablet formulation was manufactured by P.T. Kimia Farma Tbk and the comparator product was Movi-Cox® 15mg tablet, manufactured by PT. Boehringer Ingelheim Indonesia.
After 2hours, water was provided. No food was allowed until 4hours after drug administration. Standard meals were reserved for 4hours (lunch), 8hours (snack) and 12hours (dinner) after study drug administration. Subjects were remaining in a sitting position until 4hours period after drug administration. The clinical facility did not permit subjects to exit. Blood Samples Collection Blood samples were taken at specific time points to represent the drug absorption, distribution, and elimination phases. Most drugs require 12-18 blood samples, including one sample before dosing (t1), 2-3 samples before Cmax, 4-6 samples around Cmax, and 5-8 samples after Cmax [1]. In the Meloxicam test, blood samples were taken for 72hours. 5ml of blood was collected at the following time points: 0 hours (before drug administration); at 1; 2; 3; 3,5; 4; 4,5; 5; 5,5; 6; 8; 10; 12; 16; 24; 36; 48; and 72hours after drug administration (18 points). Every deviation at the time of blood collection is recorded in the CRF as a protocol deviation.
The blood samples are collected using a syringe and transferred into CPDA blood collection tubes. Plasma is centrifuged at 3000rpm for 10minutes and immediately transferred plasma into tubes. Then, the plasma is stored in the freezer at a maximum temperature of -20°C until Meloxicam analysis is conducted. The total amount of blood collected from each subject was 195ml (including 15ml for initial health screening).
Analytical Method
The analytical method used was validated according to EMEA Guideline who have met the requirements for parameter selectivity; carry-over effect; calibration curve and Lower Limit of Quantification (LLOQ); precision and accuracy; matrix effect; dilution integrity; and stabilities (i.e., freeze-thawed stability, short-term stability at room temperature, post preparative stability/autosampler batch integrity, stock solution and internal standard stability, and long-term stability) [11]. The anticoagulant used during the validation and bioanalytical phase was CPDA (Citrate Phosphate Dextrose Adenine).
LLOQ determination of the drug Meloxicam after oral administration at a dose of 15mg obtained a Cmax value is 1,023 ± 0.4102µg/ml or the equivalent of 612.8ng/ml [5]. Based on EMEA, LLOQ determination is 1/20 of Cmax [11]. The validated LLOQ data is 10ng/ml. The concentration of the Meloxicam in plasma was analysed using LCMS/MS method in the Equitrust Laboratory (Jakarta, Indonesia). Piroxicam will be used as the internal standard. Sample separation in chromatography using Acquity BEH C18column (50x2.1mm, 1.7µm).
The mobile phase used was acetonitrile and 0.2% formic acid using the isocratic system. Samples were prepared using a Protein Precipitation Extraction method. Where the supernatant which is obtained was removed into vials and injected into the LCMS/MS system. During the bioanalytical phase of the plasma samples, the analysis was monitored according to the quality control process, which included system suitability test, linearity of calibration curve, and quality control samples (Low QC, Medium QC, and High QC) referred to EMEA guideline [11].
**Pharmacokinetics Parameter**
Pharmacokinetic parameters were determined based on first-order kinetics calculations and an open one-compartment model. The following parameters were calculated for each subject during each period: AUC0-t, AUC0-inf, Cmax, tmax and t½. Pharmacokinetic analysis was performed for Meloxicam concentrations in plasma before (0hours) and up to t-hours (72hours) after drug administration. The area under the curve (AUC0-t) is calculated based on the sum of the trapezium areas formed from the relationship between levels versus time. AUC0-inf is calculated based on the equation AUC0-t + Cpt (final)/K. The elimination speed constant (K) is determined based on the slope (gradient) value of the linear line (lnCpt = ln Cpo-Kt or logCpt = logCpo - K/2,303.t). The elimination half-life (t½) is determined by the equation t½ = ln 2/K or 0.693/K. Maximum concentration (Cmax) and maximum time (tmax) are taken directly from the data showing the maximum value.
**Statistical Analysis**
Bioequivalence between the test product and the comparator is determined based on the parameters: Cmax and AUC0-t based on the average ratio with the 90% Confidence Interval (90% CI) of log data or in data. The log or ln Cmax and AUC0-t values of the two products were analysed using two-way Analysis of Variance (ANOVA). Pharmacokinetic analysis was carried out using the Ms Program.
Excel and statistical analysis using the R program. Compared drug products (Test and Comparator), drug administration period (I and II), subjects, and sequence (TR and RT) Differences in mean values of Cmax and AUC0-t between test products (test=T) and a comparison product (Comparator=R) that meets the bioequivalence criteria, namely the ratio of geometric mean values (AUC)T/(AUC)R= 1.00 with 90% CI= 80.00–125.00% (α: 0.05) and (Cmax)T/(Cmax)R= 1.00 with 90% CI= 80.00–125.00% (α: 0.05). Tmax between the two products was analysed using non-parametric methods (Wilcoxon). Meanwhile, Tmax and t½ between the two products were analysed by descriptive statistics. 90% Confidence Interval (90% CI) is calculated using the equation:
$$\frac{90\%CI}{diff.} = \frac{Difference \pm t}{0.1(n-2)} \times SEdiff.$$
$$Difference = averagelnT - averagelnR$$
$$n = number of subjects$$
$$\alpha = 0.05$$
$$SEdiff = \sqrt{MSResidualx2/n}$$
$$\frac{90\%CI}{ratio} = anti\ln \frac{90\%CI}{diff} \times 100\%$$
**Results**
Several subjects reported adverse events, namely abdominal pain, headaches and constipation after administering the test drug or comparison drug. However, not all subjects experience this (depending on the sensitivity of each subject) and this incident is not disturbing and does not have a significant effect on the subject's health. These adverse events were recorded in the CRF and the deviation during the study is reported in protocol deviation point at full study report.
Based on bioanalytical result, the pharmacokinetic parameters (AUC0-t, AUC0-inf, Cmax, t½, and tmax) of the test drug and comparator drug were calculated and compared to assessed bioequivalence. The calculated 90% CI with a = 5.00% for geometric mean of individual and the ratios of AUC0-inf and AUC0-t as well as Cmax for the test drug Meloxicam and comparator drug were all within 80.00
- 125.00% interval. This was in conformity with the standard guideline for bioequivalence study [1].
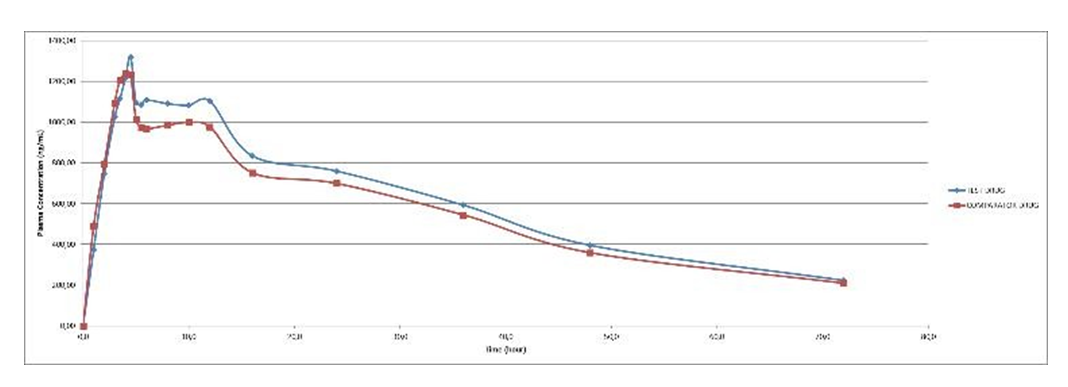
The Summary of pharmacokinetic parameters of the study shown in Table 3. Meanwhile, the main statistical calculations for AUC0-t and Cmax parameter of study Meloxicam was obtained from 18 subjects after oral administration of the test drug and comparator drug shown in Table 4. The means of plasma concentration vs. time profiles after a single dose of oral administration of test drug are shown in Figures
| Parameter | Test Drug | Comparator Drug | ||
|---|---|---|---|---|
| Arithmetic Mean | Standard Deviation | Arithmetic Mean | Standard Deviation | |
| AUC -72 0 (ng.h.ml-1) | 43,353.86 | 12,105.29 | 40,032.91 | 10,911.93 |
| AUC -inf 0 (ng.h.ml-1) | 54,337.05 | 21,890.21 | 50,724.54 | 20,005.34 |
| Cmax (ng.ml-1) | 1,402.29 | 372.57 | 1,337.20 | 398.82 |
| t½ (h) | 28.23 | 10.15 | 29.18 | 12.11 |
| tmax (h)* | 4.61 | 2.19 | 4.5 | 1.97 |
Table 3: The Summary of pharmacokinetic parameters.
| Parameter | % Ratio of Geometric Means (T/R) | 90% Confidence Interval (T/R) | ||
|---|---|---|---|---|
| Lower Limit | Upper Limit | % CV | ||
| AUC0-72 | 108.07 | 104.88 | 111.35 | 5.15 |
| Cmax | 105.24 | 97.72 | 113.34 | 12.8 |
Table 4: Statistical calculations for AUC0-72 and Cmax parameter of Meloxicam after a single-dose Oral Administration of Test and
Discussion
The objective of this study is to compare the efficacy between the generic drug of Meloxicam 15mg Tablets produced by PT. Kimia Farma Tbk and its comparator (Movi- Cox® 15mg Tablet by PT. Boehringer Ingelheim Indonesia) by a study of bioequivalence in Indonesian healthy volunteers. The study was evaluated in 18enrolled volunteers with a randomized, single-dose, open-label, two-way crossover design (2treatments, 2periods, and 2sequences) with a 7-day washout period, in a fasting state.
The primary parameters to assess the bioequivalence between the test and comparator drug of Meloxicam are AUC0-72 and Cmax. Based on the bioequivalence criteria of a 90% confidence interval, the ratio of the geometric mean values.
(AUC)T/(AUC)R=1.00 with 90% CI=(80-125)% (α:0.05) and (Cmax)T/(Cmax)R=1.00 with 90% CI=(80-125)% (α: 0.05) were used.
The geometric mean ratios (90% confidence intervals) of the test drug/ comparator drug for Meloxicam were 108,07% (104,88-111,35) for AUC0-72 and 105,24% (97,72- 113,34) for Cmax. Based on the AUC0-72 and Cmax values, Meloxicam 15mg Tablet manufactured by PT. Kimia Farma tbk is bioequivalent to Movi-Cox® 15mg Tablet manufactured by Boehringer Ingelheim Indonesia. In this study, the intra- subject coefficient of variation (%CV) obtained from ANOVA was 5.15% for AUC0-72 and 12.80% for Cmax.
Conclusion
Based on the results of this study, it can be concluded that Meloxicam 15mg Tablet produced by PT. Kimia Farma Tbk (Test) is bioequivalent to Movi-Cox® 15mg Tablet (comparator) Produced by PT. Boehringer Ingelheim Indonesia. Thus, it can be assumed that the two formulations were therapeutically equivalent with the same clinical efficacy and safety.
References
-
Badan POM (2015) Bioequivalence Study Guidelines. Jakarta BPOM.
-
American Society of Health-System Pharmacist (2011) AHFS Drug Information. USA American Society of Health- System Pharmacist.
-
Mobic-Meloxicam (2021) Connective Rx.
-
RI POM Agency (2014) Indonesian National Drug Informatorium (IONI-2017). National Library of the Republic of Indonesia.
-
Ahmad M, Ghulam M, Naveed A, Faryal S, Shujaat A (2011) Bioequivalence Study of Two Brands of Meloxicam Tablets in Healthy Human Pakistani Male Subjects. Acta Poloniae Pharmaceutica Drug Research 68(1): 115-119.
-
Meloxicam Dosage and Drug Information (2021) MIMS.
-
Gschwend MH, Aydin E, Wolfgang M, Uygur T, Ilker K, et al. (2007) Pharmacokinetic and Bioequivalence Study of Meloxicam Tablets in Healthy Male Subjects. Arzneimittel-Forschung Drug Research 57(5): 264-268.
-
Jiménez GM, José A, Alionka P, Contreras L, García A, et al. (2005) Bioequivalence Evaluation of Two Brands of Meloxicam Tablets (Promotion® and Mobicox®): Pharmacokinetics in a Healthy Female Mexican Population. Biopharm Drug Dispos 26(5): 167-171.
-
Tangsucharit P, Kampan J, Kanjanawart S, Gaysonsiri D, Vannaprasaht S, et al. (2009) Bioequivalence Study of Two Meloxicam Tablet Formulations After Single- dose Administration in Healthy Thai Male Volunteers. Int J Clin Pharmacol Ther 47(10): 638-642.
-
Bioequivalence-General Considerations (2016) World Health Organization.
-
Guideline on Bioanalytical Method Validation (2011) European Medicines Agency (EMEA).
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State