Effect of Crospovidone, Croscaramellose Sodium in Combination on the Drug Release of Anti diabetic Medication in Tablet Form
Objective: The current study’s objective is to check the impact of superdisintegrants on the drug release of Vildagliptin by developing oral disintegrating tablets (ODT). Vildagliptin, new class of oral hypoglycemic agent. It acts by inhibiting dipeptidyl peptidase-4. Conventional Vildagliptin tablets are (due to its first pass effect) unsuitable where quicker onset of action is required Hence, it is necessary to develop oral disintegrating tablet, in order to obtain improved patient compliance. Methods: Using Oral disintegrating tablets of Vildagliptin were prepared using various quantities of Crospovidone & Croscaramellose sodium as Superdisintegrants by direct compression method. Nine trials were developed by varying the quantities of Crospovidone, Croscaramellose sodium such as 6 ± 1.5 mg i.e. 4.5, 6, 7.5 mg respectively. Prepared formulations were assessed for Pharmaceutical Product Performance as per official methods such as hardness, friability, weight variation, drug content, wetting time, disintegration time, and drug release studies. Results: Findings indicate that all formulations meet the acceptance criteria such as good mechanical strength (>4.3 kg/cm2), less friable (<0.65%), uniform drug content (>98%) and kinetic modeling was applied to the in-vitro dissolution profiles. Conclusion: The best formulation (F1), which is likewise identical to the marketed product (Galvus-50) (f2= 89.01, f1= 1.85), contained 7.5 mg of Crospovidone and 7.5 mg of Croscaramellose sodium. Formulation (F1) follow first order, whereas release mechanism found to be Fickian type (n= 0.323).
Introduction
The pharmaceutical market gives Oral Disintegrating Tablets (ODT) a unique place. ODT was regularly replaced with melt-in-the-mouth pills and Fast dissolving tablets, Oral/ Mouth dissolving Tablets [1]. Rapid disintegrating tablets can be readily available for disintegration, they breakdown in the mouth within 60 seconds. Based on the manufacturing process, they show changes in typical organoleptic features including masking sweetness or taste and better palatability. Additionally, they show changes in quality control metrics like breaking index, drug release from formulation, stability, and clinical result. FDTs can be prepared using a variety of procedures, some of which are cotton candy process, granulation techniques, named technologies (Durasolv, Orosolv), spray drying, trituration, moulding, lyophilization/ freeze drying, and mass extrusion [2].
Vildagliptin is a dipeptidyl peptidase-4 (DPP4) inhibitor that is the new class of oral hypoglycaemic agent. By such inhibition of DPP-4 enzyme it prevents the glucose- dependent insulin tropic polypeptide (GIP) and Glucogen- like peptide-1 (GLP-1), the incretic hormone degradations. Conventional Vildagliptin tablets available in the market are not suitable (due to its extensive first pass metabolism) where quick onset of action is required. Hence an effort was made to formulate it as the oral disintegrating tablet to boost its bioavailability (by bypass the first pass metabolism) [3, 4, 5].
An attempt was made to maximize the drug delivery from formulation with the help of combination super disintegrates at various concentrations (Crospovidone, Croscaramellose sodium) by formulating the Oro dispersible tablets for Vildagliptin. Tablets by Direct Compression Techniques have a Unique Nature in the Form of Less Time Consumption, Rapid Production, and Economy in the Operational Management among the Many Methods of Manufacturing Techniques Available [2, 6].
Materials and Methods
Materials
Vildagliptin was a gift sample procured from Vensa Pharma Pvt Ltd, India. Microcrystalline Cellulose, Croscaramellose sodium, Crospovidone were procured from National Scientifics, Guntur. Other excipients were procured from High Chemie Ltd, Vadodara.
Preparation of Vildagliptin Oral Disintegrating Tablets
The direct compression approach was used in the production of Vildagliptin ODT as per the Formulae shown in Table 1. All of the components were sifted using 40 meshes (#40) to produce a uniform fine blend. Lubricants were screened through #60, combined with the mixture above, and mixed well. These blends were subjected to compression to produce ODT using a tablet minipress (8 stations) and circular punches that measure 8 mm in diameter. In-Process Quality Control (IPQC) tests were performed on the acquired tablets. For storage and subsequent processing, finished tablets were transferred to airtight, light-resistant containers (Table 1) [7].
| Name of Ingredients | Quantity of Ingredients per each Tablet (mg) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| F 1 | F 2 | F 3 | F 4 | F 5 | F 6 | F 7 | F 8 | F 9 | |
| Vildagliptin | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Micro Crystalline Cellulose (MCC) | 40.5 | 41.25 | 42 | 41.25 | 42 | 42.75 | 42 | 42.75 | 43.5 |
| Mannitol | 40.5 | 41.25 | 42 | 41.25 | 42 | 42.75 | 42 | 42.75 | 43.5 |
| Crospovidone | 7.5 | 7.5 | 7.5 | 6 | 6 | 6 | 4.5 | 4.5 | 4.5 |
| Croscaramellose Sodium | 7.5 | 6 | 4.5 | 7.5 | 6 | 4.5 | 7.5 | 6 | 4.5 |
| Talc | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Magnesium Stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Total Weight | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 |
Table 1: Formulae for the Development of Vildagliptin Oral Disintegrating Tablets.
Evaluation of Vildagliptin Oral Disintegrating Tablets Hardness: It was carried out with the help of Monsanto Tablet Hardness Tester. The diametric breakage of tabled when placed between two jaws of apparatus is considered as mechanical strength or breaking index [8]. Friability/ Durability: Twenty tablets were weighed and noted as W0 cumulatively (Initial weight). The pills were then dedusted with a Roche Friabilator for 4 minutes at a speed of 25 rpm, and weighed again recorded as (W). The following equation was used to obtain the percentage of friability (%Friability ≤1).
Assay: 20 tablets were chosen and ground in an impartial manner. The powder corresponding to 100 mg of Vildagliptin was weighed, added to a 100 mL volumetric flask with 60 mL of Phosphate Buffer Solution (PBS) pH 6.8, and then sonicated for 10 minutes to completely solubilize the medication. The resulting solution was then diluted with PBS pH 6.8 to make up the required volume. Prepare a further 2 mL aliquot from that for dilution in 100 mL of PBS pH 6.8. Using a UV-visible spectrophotometer, the resulting solution was analyzed for its absorbance at 276 nm. Thickness: It was measured with the help of verinier calipers. To measure the tablet thickness simply place the tablet in between the jaws and slide the scale jaw to press the tablet against the stationary jaw, look at the number on the main scale that lines up with the sliding scale’s zero. Then, find the mark on the Vernier scale that line up with a number on the main scale. Finally, add those two numbers together to get your measurement [9]. Wetting time: Tablets were placed on a petridish containing paper that had been soaked in 5mL of distilled water to measure the wetting time of the tablets. The tablet’s wetting time was measured in seconds. In-vitro Dissolution Study: Vildagliptin ODT was analyzed for drug release study utilizing a Lab-India USP type-II tablet dissolution test apparatus and 900 ml of PBS pH 6.8 buffers in accordance with the recommended method as outlined in the monograph. Using a UV-visible spectrophotometer, samples’ absorbance was measured at 276 nm and the data was subjected to kinetic modeling [10, 11].
Disintegration test: According to the guidelines of the modified disintegration test for tablets, this test was conducted. Only 2 ml of medium were allowed to fall below the sieve in a cylindrical cylinder with 10#. Time of disintegration was noted [12].
Results and Discussion
9 different formulations of Vildagliptin oral disintegrating tablets were prepared utilizing the direct compression method using varying ratios of super disintegrates in accordance with the formulae shown in Table 1. Pharmaceutical product performance tests were conducted on the developed formulations. Table 2 displayed the information.
All tablets were discovered to be less brittle and to have acceptable mechanical strength. Hardness for all the formulations was found with in the range of 4.2± 0.13 to 4.4 ± 0.06 Kg/cm2. The lower standard deviation value indicates that the hardness of all the formulations was almost uniform and possesses good mechanical strength with sufficient hardness. The Thickness values were found in the range from 4.45 ± 0.004 mm to 4.7 ± 0.055 mm. Uniformity in the values indicates that formulations were compressed without sticking to the dies and punches. The study results for friability were found well within the approved range (<0.7%) in all the formulation. Results revealed that the tablets possess good mechanical strength. The produced tablets’ uniformity of weight and drug content were both within acceptable ranges. Average weight for all formulations was found to be in the range of 149±0.53 to 150.55±0.5 mg. The weight of all the tablets was found to be uniform. This is due to good flow property and compressibility of all the formulations. The Drug content test also reveals that all formulation was founded to be within the Pharmacopoeial limit of ±10%. It was founded to be from 98.02±0.55 to 99.88±0.7 mg. This reveals all formulations uniform (Table 2).
| Hardness (kg/cm2) | Thickness (mm) | Friability (%) | Average Weight | Drug Content (%) | Wetting Time (sec) | Disintegration Time (sec) | |
|---|---|---|---|---|---|---|---|
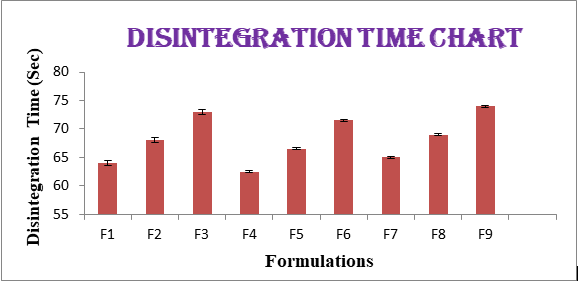
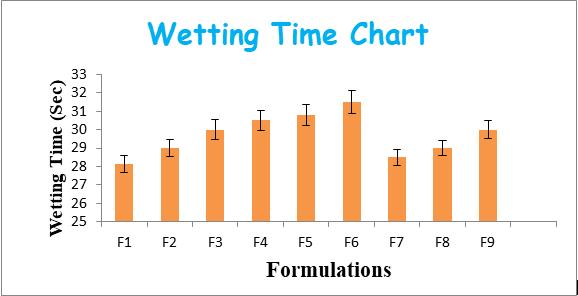
| F1 | 4.4±0.057 | 4.7±0.053 | 0.6±0.01 | 150.55±0.46 | 99.88±0.703 | 28.12±0.46 | 64±0.394 |
| F2 | 4.35±0.105 | 4.65±0.0525 | 0.63±0.015 | 149.3±0.94 | 99.23±0.7245 | 29±0.454 | 68±0.441 |
| F3 | 4.3±0.104 | 4.7±0.055 | 0.645±0.013 | 149.45±0.56 | 98.55±0.575 | 30±0.531 | 73±0.418 |
| F4 | 4.35±0.057 | 4.5±0.041 | 0.57±0.007 | 150.25±1.09 | 99.42±0.788 | 30.5±0.563 | 62.5±0.155 |
| F5 | 4.3±0.105 | 4.45±0.04 | 0.6±0.012 | 149±0.53 | 98.77±0.81 | 30.8±0.557 | 66.5±0.203 |
| F6 | 4.25±0.104 | 4.5±0.043 | 0.615±0.01 | 149.15±0.90 | 98.08±0.66 | 31.5±0.634 | 71.5±0.181 |
| F7 | 4.3±0.079 | 4.7±0.041 | 0.56±0.01 | 150.90±0.75 | 99.35±0.678 | 28.5±0.424 | 65±0.175 |
| F8 | 4.25±0.126 | 4.65±0.042 | 0.59±0.015 | 149.65±0.19 | 98.70±0.699 | 29±0.418 | 69±0.223 |
| F9 | 4.2±0.126 | 4.7±0.044 | 0.61±0.013 | 149.8±0.45 | 98.02±0.549 | 30±0.495 | 74±0.2 |
Table 2: Post-Compression Parameters for Vildagliptin Oral Disintegrating Tablets.
The rationality for the total 9 Formulations, F1 to F9 use of two different Superdisintegrants in different compositions from High–High to Low-Low concentrations shows variable results in all post compression parameters, compared with pure drug all were improved solubility and dissolution due to lesser disintegration and other parameters. The faster disintegration obtained with high concentrations of Crospovidone and Croscaramellose sodium mainly because, Crospovidone uses a combination of swelling, wicking and deformation mechanism for rapid disintegration of tablets, swells rapidly in water without forming gel. Whereas Croscaramellose sodium works by combination of swelling and wicking mechanism for disintegration. Hence combination showed rapid disintegration. When the concentration of Crospovidone increases alone there is a chance to friable more, that’s the reason given in combination to maintain all physical properties and withstand the effects like wear and tear.
The effect of combination of super disintegrates in low concentrations shows more disintegration time which is undesirable or not having a significant use of combination. All the formulations showed wetting time in the range of 28.12±0.46 to 30±0.5 sec. All the formulations showed Disintegration time in the range of 64±0.394 to 74±0.2 sec and the same was represented as Figure 1,2. From the results of wetting time and Disintegration time, reveals that as the concentration of superdisintegrants increases the wetting time decreases (Concentration of superdisintegrants inversely proportional to wetting time). Finally, formulation F1 showing promising results for made as ODT (Figures 1 & 2).
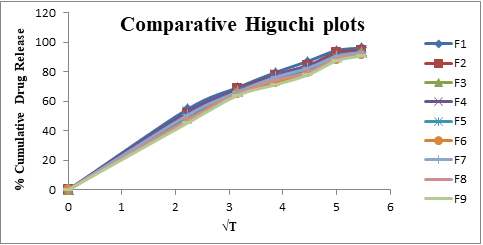
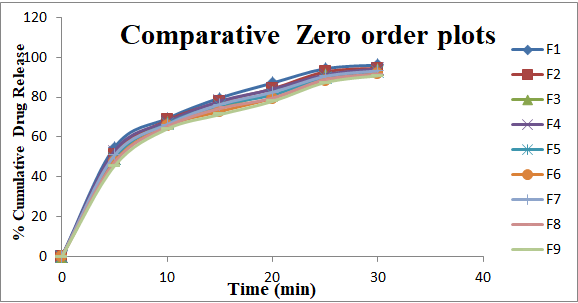
From the above results reveals that as the concentration of super disintegrates increases dissolution also improved (due to improved disintegration). From Formulation F1 to F9, the superdisintegrants combination was varied to get optimum concentrations for both the superdisintegrants to get desirable effect for showing promising result comparable to marketed Formulation. Dissolution profiles of Vildagliptin oral disintegrating tablets were well fit to kinetic modeling, results presented in Table 3 and the same was shown in Figures 3-6.
The combined effect of various composition of Crospovidone and Croscaramellose sodium on the Vildagliptin release were studied using response surface plots (RSM) shown in Figure 7-11. RSM plots were constructed using Sigma plot V13 (Figures 7-11) (Table 3,4).
| S.NO | Formulation Code | Statistical Parameters | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero order | First order | Higuchi | Korsmeyer-Peppas | ||||||||||
| a | b | r | a | b | r | a | b | r | a | n | r | ||
| 1 | F 1 | 27.482 | 2.751 | 0.884 | 1.959 | 0.046 | 0.994 | 7.953 | 17.569 | 0.983 | 1.517 | 0.323 | 0.998 |
| 2 | F 2 | 25.998 | 2.73 | 0.892 | 1.948 | 0.041 | 0.993 | 6.955 | 17.337 | 0.985 | 1.479 | 0.345 | 0.996 |
| 3 | F 3 | 24.795 | 2.713 | 0.899 | 1.951 | 0.038 | 0.993 | 6.216 | 17.132 | 0.988 | 1.454 | 0.357 | 0.996 |
| 4 | F 4 | 26.646 | 2.682 | 0.885 | 1.935 | 0.039 | 0.994 | 7.654 | 17.116 | 0.983 | 1.502 | 0.325 | 0.998 |
| 5 | F 5 | 25.163 | 2.661 | 0.893 | 1.933 | 0.036 | 0.992 | 6.657 | 16.884 | 0.986 | 1.463 | 0.348 | 0.995 |
| 6 | F 6 | 23.959 | 2.645 | 0.901 | 1.938 | 0.034 | 0.992 | 5.917 | 16.679 | 0.988 | 1.437 | 0.36 | 0.995 |
| 7 | F 7 | 25.466 | 2.679 | 0.893 | 1.937 | 0.037 | 0.994 | 6.825 | 17.001 | 0.986 | 1.473 | 0.342 | 0.997 |
| 8 | F 8 | 23.982 | 2.658 | 0.9 | 1.937 | 0.034 | 0.992 | 5.827 | 16.769 | 0.988 | 1.432 | 0.366 | 0.994 |
| 9 | F 9 | 22.779 | 2.641 | 0.908 | 1.943 | 0.033 | 0.992 | 5.088 | 16.564 | 0.99 | 1.405 | 0.38 | 0.994 |
Table 3: Statistical Parameters for kinetic modeling.
| S.NO | Formulation Code | Kinetic Parameters | ||||
|---|---|---|---|---|---|---|
| t (Min) 10% | t (Min) 1/2 | t (Min) 90% | Wetting Time (Sec) | Disintegration Time (Sec) | ||
| 1 | F 1 | 6.581 | 13.161 | 21.867 | 28.12±0.46 | 64±0.394 |
| 2 | F 2 | 7.353 | 14.705 | 24.433 | 29±0.454 | 68±0.441 |
| 3 | F 3 | 7.823 | 15.645 | 25.995 | 30±0.531 | 73±0.418 |
| 4 | F 4 | 7.688 | 15.375 | 25.546 | 30.5±0.563 | 62.5±0.155 |
| 5 | F 5 | 8.394 | 16.787 | 27.892 | 30.8±0.557 | 66.5±0.203 |
| 6 | F 6 | 8.833 | 17.665 | 29.351 | 31.5±0.634 | 71.5±0.181 |
| 7 | F 7 | 8.086 | 16.173 | 26.871 | 28.5±0.424 | 65±0.175 |
| 8 | F 8 | 8.766 | 17.532 | 29.13 | 29±0.418 | 69±0.223 |
| 9 | F 9 | 9.195 | 18.39 | 30.556 | 30±0.495 | 74±0.2 |
Table 4: Results for Response variables.
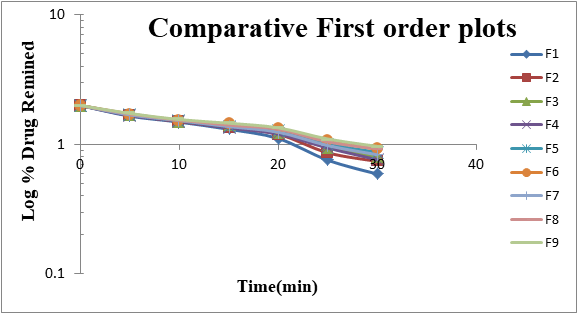
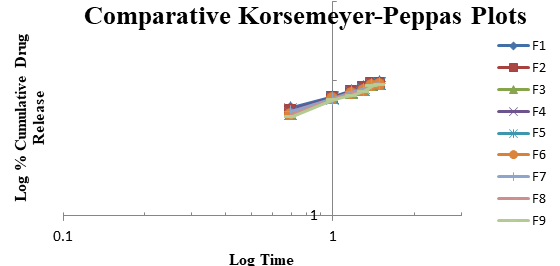
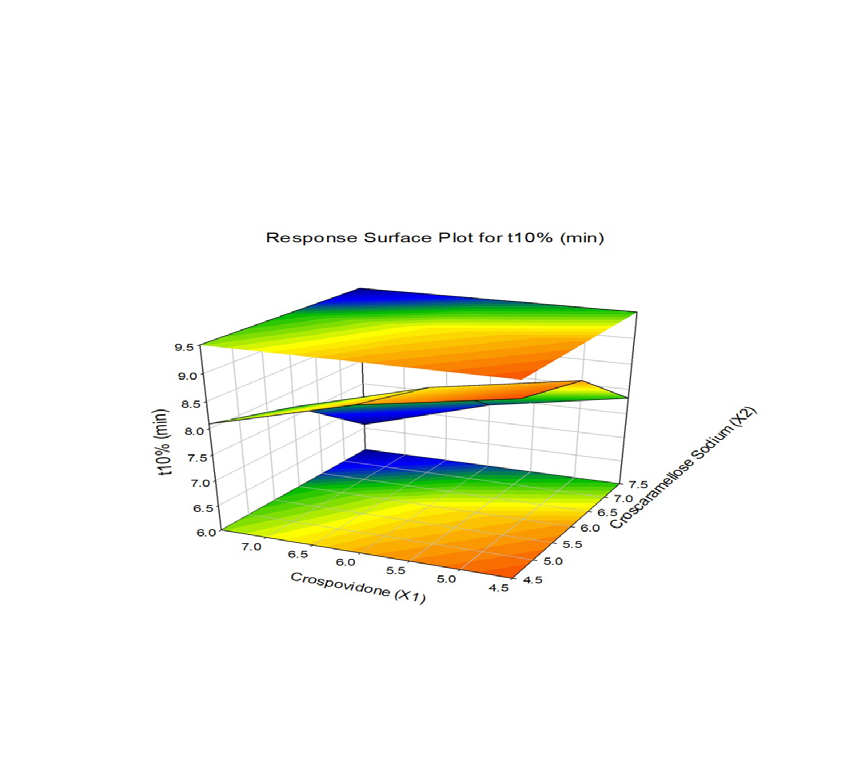
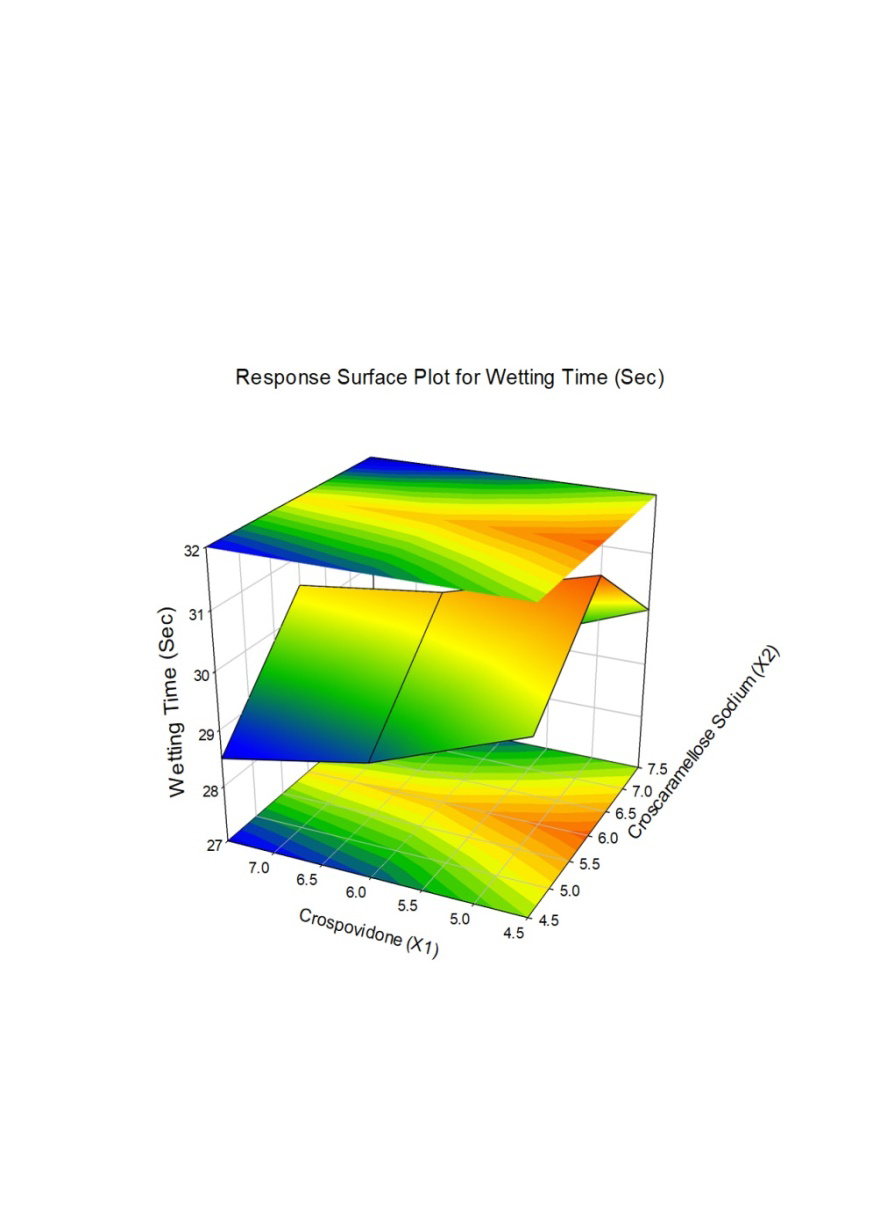
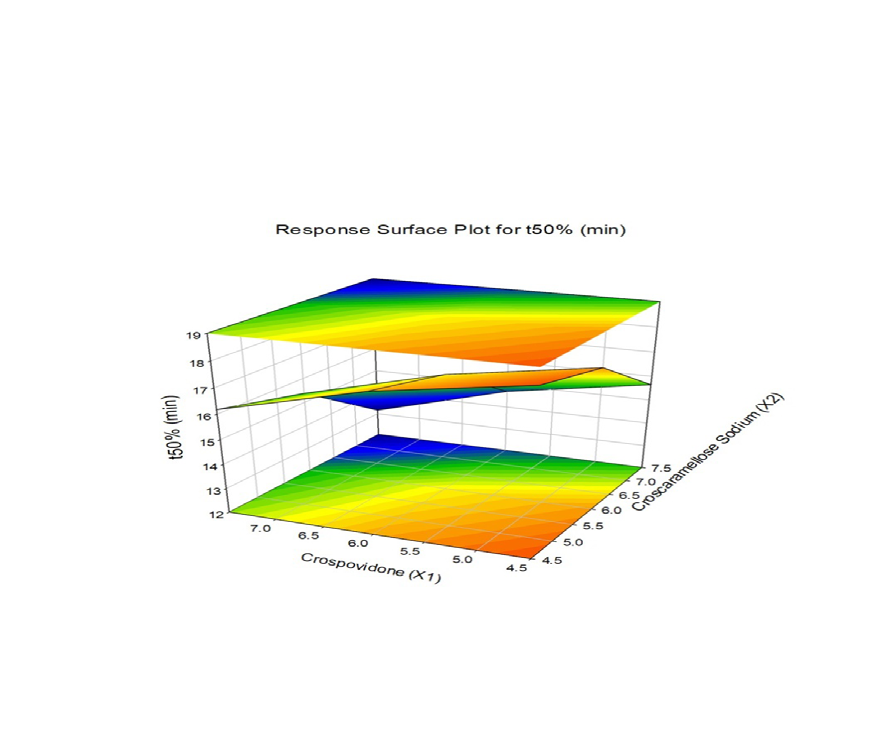
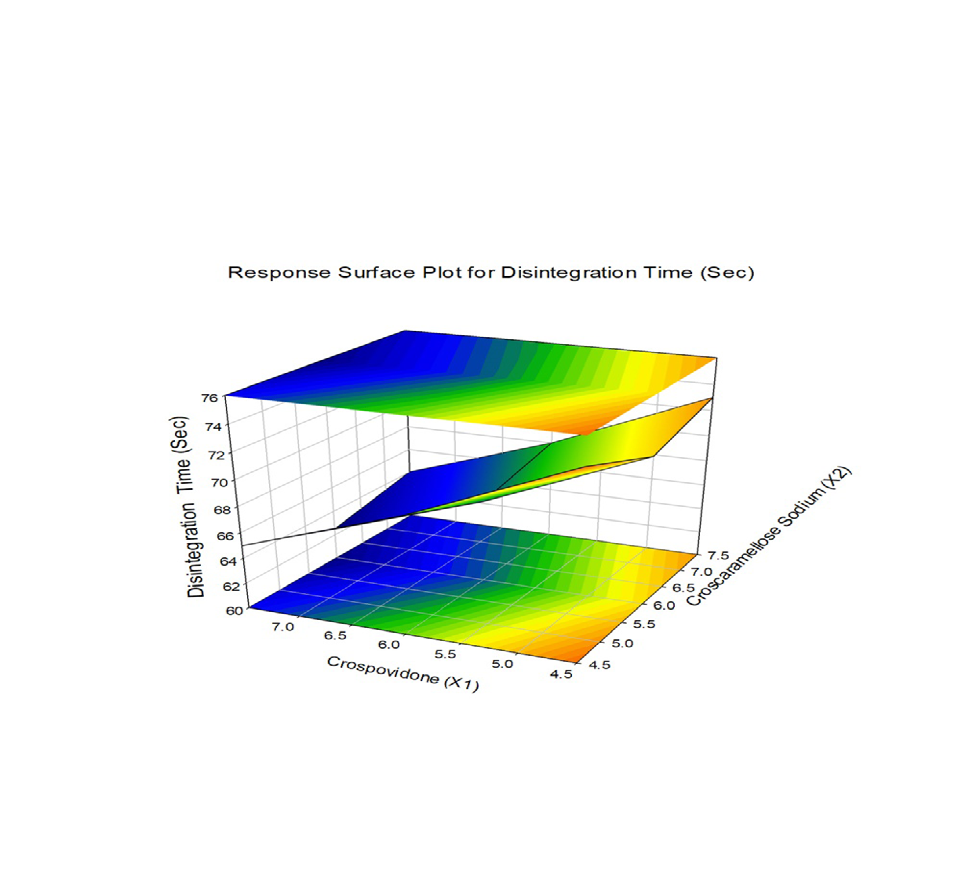
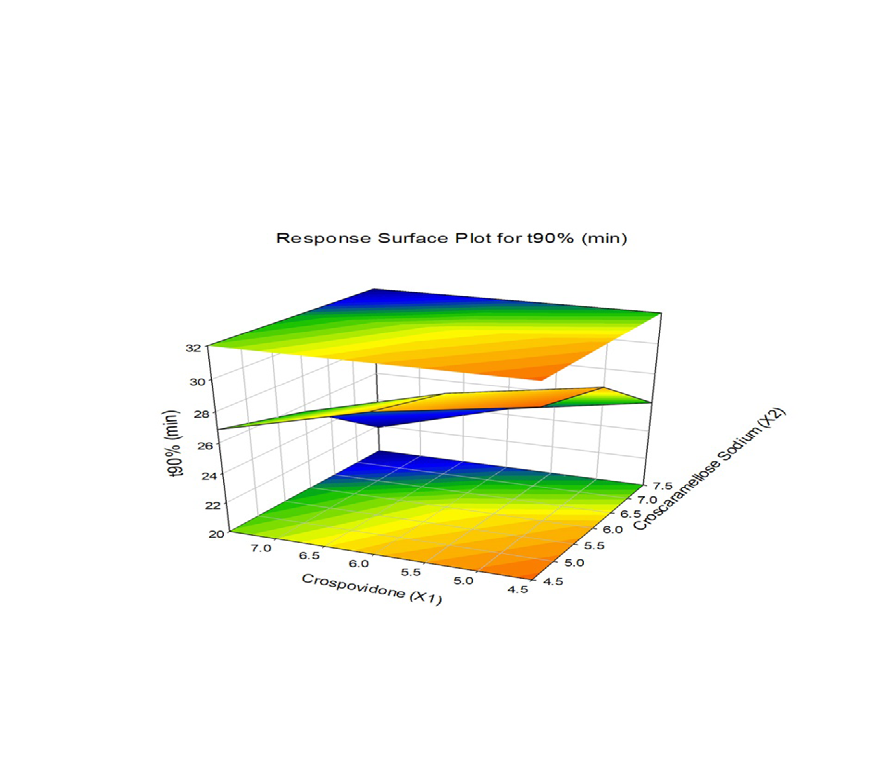
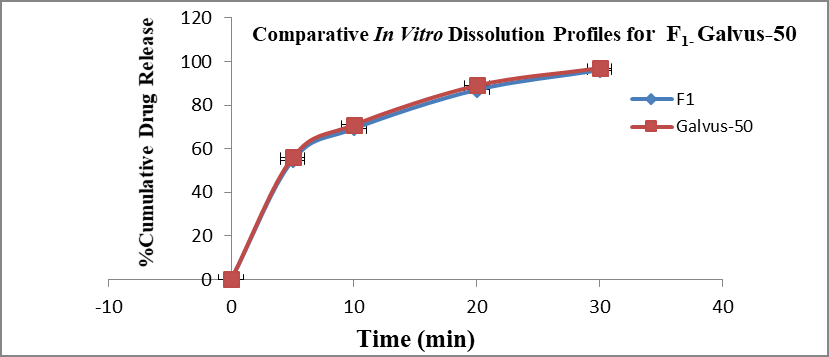
F1 is regarded as the best formulation among all batches (based on Desirability). F1, which contained 7.5 mg of Croscaramellose sodium and Crospovidone in equal amounts, produced promising dissolution characteristics that aid in achieving the goal of the study through faster disintegration and rapid dissolution. Table 4 provides a summary of the data for the derived kinetic parameters. The in-vitro dissolution profile of F5 was compared with Marketed product (Galvus-50) tablets, shows similarity f2= 89.01; f1= 1.85 and the same was presented as (Figure 12).
Conclusion
The current study focuses on the impact of using superdisintegrants for the development of Vildagliptin ODT, such as Croscaramellose sodium and Crospovidone. F1 follows first order type of kinetics, higuchi type model whereas the mechanism of drug release follows Fickian diffusion. The best formulation F1 may be used for the effective management of Type-II Diabetes mellitus also contributes to reduced hyperglycaemia and assists in weight loss and reduction of blood pressure.
Acknowledgements
The management and staff of the Narasaraopeta Institute of Pharmaceutical Sciences in India are gratefully acknowledged by the authors for the facilities provided and ongoing support for the completion of the current study.
References
-
Gunda RK, Kumar JNS (2018) Formulation Development and Evaluation of Amisulpride Fast Dissolving Tablets. FABAD J Pharm Sci 43(2): 105-115.
-
Gunda RK, Kumar JNS, Satyanarayana V, Swaruparani G, Prasad BS (2016) Formulation Development and Evaluation of Clopidogrel Fast Dissolving Tablets. Iranian Journal of Pharmaceutical Sciences 12(2): 61-74.
-
Chithira HB, Sathyaraj A, Karpagavalli L, Roosewelt C (2023) Formulation and evaluation of oral disintegrating tablets of Vildagliptin. World Journal of Pharmacy and Pharmaceutical Sciences 12(9): 2108-2134.
-
Chadha M, Nautiyal U (2023) Formulation and evaluation of anti-diabetic tablet of vildagliptin and effect of disintegrants concentration on drug release. YMER 22(3): 711-720.
-
Narasimha rao B, Lakshminarasappa DK, Viswanath V, Gnana Prakash K, Rahath Fathima S (2016) Formulation and in vitro characterisation of vildagliptin fast disintigrating tablets. Journal of Global Trends Pharmaceutical Sciences 7(3): 3374-3381.
-
Gunda RK, Manchineni PR, Kotcherla AK, Dhachinamoorthi D, Koteswara Rao G (2021) The Effect of pH dependent and pH independent polymers on the drug release of anti-ischemic agent. International Pharmacy Acta 4(1): 1-6.
-
Gunda RK, Kumar JNS (2017) Formulation Development and Evaluation of Moxifloxacin: Hcl Fast Dissolving Tablets. Pharmaceutical Methods 8(2): 160-167.
-
Gunda RK, Manchineni PR (2020) Statistical Design and Optimization of Sustained Release Formulations of Pravastatin. Turk J Pharm Sci 17(2): 221-227.
-
Gunda RK, Vijayalakshmi A, Masilamani K (2020) Development, In-vitro and In-vivo evaluation of gastro retentive formulations for Moxifloxacin HCl. Research Journal Pharmacy Technology 13(10): 4668-4674.
-
Higuchi T (1963) Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci 52: 1145-1149.
-
Peppas NA (1985) Analysis of Fickian and non-fickian drug release from polymers. Pharm Acta Helv 60(4): 110-111.
-
Gunda RK, Kumar JNS, Satyanarayana V (1985) Formulation Development and Evaluation of Carbamazepine Fast Dissolving Tablets. J Pharm Res 10(5): 216-225.
- Hydrogen Peroxide Scavenging by Methanolic Extracts of Coriander: An In Vitro Antioxidant Study
- Aromatherapy in Palliative Care: A Fragrant Quest for Relief
- Empowering Women, Securing Futures: Contraception’s Role in Socioeconomic Progress in India
- Knowledge, Attitudes, Anxiety, and Preventive Behaviors Regarding Covid-19 Affliction among Healthcare Workers in Pakistan
- “Competitive Landscape and Brand Equivalents: Implications for ANDA (Abbreviated New Drug Application) Approval”
- Novel Advancements in Mouth Dissolving Film Technology