We are Reporting Bardet-Biedl Syndrome (BBS) in a Term Infant Presenting with Intrauterine Growth Retardation, Acute Respiratory Distress, Polydactyly, Bilateral Hydronephrosis and Microcephaly
Bardet-Biedl syndrome is an uncommon disorder in newborn infants. A near term, female infant presented with growth retardation, polydactyly, bilateral hydronephrosis and microcephaly. Genetic testing showed heterozygous mutations of BBS10 gene for autosomal recessive Bardet Biedl Syndrome. A pathogenic variant, c. 2119_2120del (p.Val 707) and a variant of uncertain significance, c.590>G(p.Tyr 197Cys) was identified in BBS10
Introduction
Bardet Biedl syndrome (BBS) is a genetic disorder that presents with multi system involvement and variability of initial presentation. These patients may present with obesity, retinitis pigmentosa, renal cysts and hand or foot polydactyly [1]. Multiple BBS gene mutations have been reported with this disorder.
Mutations in twenty different genes have been identified. Mutations in BBS1 to BBS18 gene accounts for about 70%–80% [2]. Major features are retinal dystrophy, obesity, polydactyly, impaired cognition, hypogonadism, renal disease and a minor feature are developmental delay, seizures, anosmia, oral and dental abnormalities, cardiac abnormalities, gastrointestinal abnormalities, and endocrinopathies [3]. Early presentation could be facial dysmorphism, polydactyly and delay in acquisition of the milestones. The typical facial features of the disorder are depressed nasal bridge, micrognathia, maxillary hypoplasia, and high arch palate.
2808 g, 39-week gestation infant was born by vaginal delivery with Apgar scores of eight at one and nine at five minutes. Mother was gravida 8, para 4344. She has had 3 spontaneous miscarriages. She was hepatitis-C antibody positive. She had hepatitis-A infection in 2nd trimester of pregnancy. She is HBS Ag negative. She has had human papilloma virus infection requiring LEEP procedure 11 years ago. She was on Subutex 8 mg twice a day during pregnancy. She has had history of methicillin-susceptible staph infection in previous 2 years. She was a former smoker. She quit smoking during this pregnancy. She denied history of alcohol use. She has had history of renal abscess in the past. Prenatal studies were suggestive of bilateral fetal echogenic kidneys and echogenic focus in the heart. Placenta was circumvallated. Her urine drug screen was positive for buprenorphine metabolites and was negative for marijuana, opiates, or amphetamine.
The pregnancy was complicated by intrauterine growth retardation. The infant required oxygen by blow at referring hospital. Chest x-ray showed right pneumothorax. Thoracentesis was performed and about twenty mlL of free air was suctioned out. Follow-up chest x-ray on admission to neonatal ICU showed small right-sided pneumothorax. The infant was transferred to neonatal intensive care unit
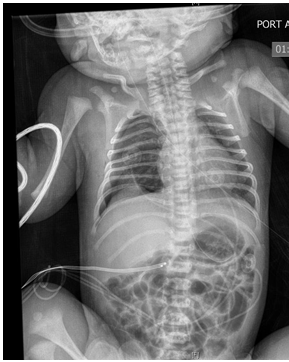
on bubble CPAP 5 cm. Initial physical exam showed weight in 17th percentile, length 25th percentile (48.3cm), and head circumference below 3rd percentile. Infant had mild tachypnea with mild substernal retractions with relatively distant heart sounds. Infant had small cleft of the soft palate. Echocardiogram showed patent foramen ovale/ atrial septal defect and left pulmonary artery stenosis. There was no evidence of facial dysmorphism. The infant had a supernumerary digit on the left foot without any bones. The infant was weaned off CPAP within 24 hours after birth and she stayed on room air subsequently. No apnea or bradycardia was noted. Renal sonogram showed right grade 1 and left grade 2 hydronephrosis. The infant was noted to have neonatal abstinence syndrome requiring morphine and clonidine. Both medications were gradually tapered. The infant was weaned off morphine on day 26 of life. Head sonogram was reported normal. Urine for cytomegalovirus was reported negative. There was no evidence of any seizure activity (Figures 1-3).
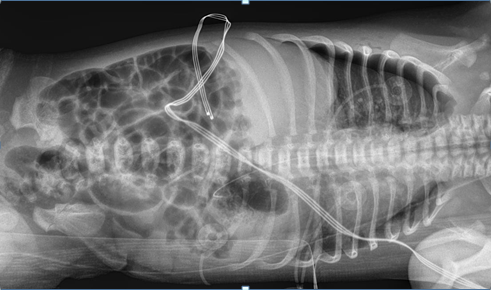
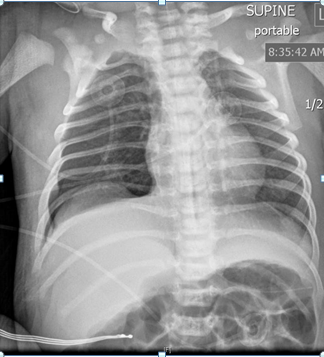
Infant was regularly evaluated by a team of physical therapists, and the infant was able to achieve full hand and digit opening, and wrist extension. She lacked full elbow extension; shoulder was lacking ~30-45 degrees of elevation. The infant grossly lacked knee extension by 5-10 degrees, hip extension lacked by 10-15 degrees from neutral position. She continued to have significant hypertonia of extremities. Infant had feeding problem during hospital stay. Gradually, nipple feedings improved and by day 28 of life she was nippling all the feedings. Karyotype was reported normal. Genetic testing showed heterozygous mutations of BBS10 gene for autosomal recessive Bardet Biedl Syndrome. A pathogenic variant, c. 2119_2120del (p.Val 707) and a variant of uncertain significance, c.590>G(p.Tyr 197Cys) was identified in BBS10. It is unknown if these variations came from the same or opposite chromosomes. Genetic testing for the mother and the family members is being arranged.
Discussion
Bardet Biedl syndrome manifests as a genetic multi- system disorder with wide variability in presentation. In 1880s, Laurence and Moon described a family with obesity, intellectual impairment, and retinitis pigmentosa. Subsequently, these patients developed spastic paraparesis. BBS disorder is seen in 1 in 100,000 in North America and Europe. Twenty-one BBS gene mutations account for majority of the clinical diagnosis of BBS. Mutations in BB 1 and BBS 10 account for majority of the genotypes in North America [2].
There are wide variations in the clinical presentation of BBS syndrome. The diagnosis may be missed in infancy and may present between 5-10 years of age with night blindness. Isolated polydactyly can present in the immediate neonatal period. Relatively common presentation of BBS syndrome is obesity. Multi organ ciliopathy presents with retinal rods and cones dystrophy, obesity, postaxial polydactyly, cognitive dysfunction, hypogonadism, high arch palate, genitourinary and renal malformations. Vision abnormalities like strabismus, astigmatism and cataracts have been reported. There could be dysmorphism, hearing loss, musculoskeletal abnormalities, hypertonicity, developmental delay, and seizures. Major features are retinal dystrophy, central obesity, polydactyly, cognitive dysfunction, genitourinary abnormalities, renal disease, and the minor features are developmental delay, seizures, anosmia, dental oral abnormalities, hepatic abnormality, endocrine will abnormalities, polycystic ovaries. Forsyth, et al. [4] suggested that the diagnosis can be made by presence of four major features or three major features and two minor features. Our index patient had a head circumference less than 3rd percentile and weight was in 25th percentile. She had echogenic kidneys on the fetal echocardiogram.
The typical facial features of the disorder are depressed nasal bridge, micrognathia, maxillary hypoplasia, and high arch palate. BBS1 mutation presents with generally less severe disease compared to other gene mutations. They have less likelihood of significant renal disease, endocrinopathies or metabolic syndrome. Retinitis pigmentosa is also of less severe degree. The rod and cones dystrophy could be delayed until seven or eight years of age. It may manifest as inability to see late in the evening. The vision progressively worsens with severity of night blindness [5]. There may be loss of peripheral vision followed by loss of central vision leading to legal blindness. Electroretinography is recommended and may show early changes within the first two years of life, although significant changes are rarely visible before the age of five [6]. Most infants will have normal birthweight and 30%
will develop obesity by age of one [7]. There is a prevalence of type two diabetes mellitus. Some of the patients will present with anosmia secondary to changes in olfactory bulb. 70% of infants with BBS present with polydactyly, commonly on the feet. There may be associated syndactyly of fingers or toes. These patients may also present with small male gonads, small penis, delay in descent of testicles into scrotum, and delay in onset of puberty. The delay in descent of testicles in these patients puts them at higher risk for testicular cancers unless orchiopexy is done at an early age. Kidney abnormalities lead to significant morbidity and mortality [8]. Some patients develop hydronephrosis of variable degree that may lead to a potential risk for pyelonephritis and renal scarring.
Learning difficulties may present with delayed speech or delay in reading skills that appears to be unrelated to visual compromise. Neurological signs could be a delay in acquisition of milestones, poor hand eye coordination, and delayed gross and fine motor skills. These patients tend to walk with the legs wide apart. Some infants with BBS may present with facial dysmorphism manifesting as hypertelorism, downward slanting of the lids, high arched palate, cleft palate, and crowding of teeth.
BBS gene is important for encoding of protein for functioning of cilia in the paranasal sinuses and rod cells in the retina. Ciliary dysfunction may lead to rod and cone dystrophy and kidney abnormalities. Lack of ability to smell, hearing deficit and situs inversus has been reported. The risk for two carrier parents to pass a gene in a child is 25%. A child is likely to have a carrier state for the illness in 50% of cases. Males and females are affected in equal numbers.
BBS needs to be differentiated from other syndromes that may have similar presentation. They are Joubert syndrome, Jejune syndrome, Sensenbrenner syndrome, Senior-Løken syndrome, McKusick-Kaufman syndrome, Laurence Moon syndrome, Alstrom syndrome, Meckel’s Gruber syndrome, Prader-Willi syndrome, and Biemond syndrome. McKusick- Kaufman syndrome is uncommon genetic disorder that presents with post axial polydactyly, congenital heart disease, genitourinary abnormalities, hypoplastic lungs, gastrointestinal abnormalities and hydronephrosis [9]. In females, it can present with hydrometrocolpos. The gene abnormalities are related to BBS6, also known as MKKS gene.
Lawrence Moon- Biedl syndrome is autosomal recessive disorder with visual deficit and associated pituitary dysfunction. It is associated with ataxia, peripheral neuropathy, hypertonia, and contractures of limbs. It manifests with paraplegia without polydactyly and obesity [10]. The disorder is commonly reported among adult population in Kuwait. There is a wide variability of intellectual deficit. Alstrom syndrome is an autosomal recessive disorder presenting with disorder of vision and hearing, risk for obesity, diabetes mellitus, and a progressive renal compromise. It presents with visual deficit and nystagmus. There may be associated cutaneous and cardiac involvement. In this condition there is no polydactyly or learning disability [11].
Meckel Gruber syndrome is an autosomal recessive disorder presenting with multi-system involvement. Occipital encephalocele, polydactyly, cystic kidneys, hepatic fibrosis, cystic dysplasia of the lungs or thyroid, retinal colobomas, and situs defects have been reported. There may be partial defects of the corpus callosum or Dandy–Walker malformation [12]. There may be mutations with BBS2, BBS4 and BBS6 gene.
Biemond syndrome type 2 is an autosomal recessive disorder presenting with coloboma of iris, polydactyly, learning disabilities, obesity, renal abnormality, and genital abnormality [13]. Prader-Willi syndrome presents with hypertonia, feeding difficulties, failure to thrive, short stature, genital abnormalities, and excessive appetite. Subsequently, these infants develop worsening obesity and developmental delay. They have behavioral disorders and compulsive behavior. The disorder is due to abnormality of genes on chromosome 15 [14].
Joubert syndrome typically has an autosomal recessive pattern of inheritance, it is rarely X-linked recessive [15]. The syndrome is related to more than 20 genes. Most present with neonatal hypotonia and ataxia in early childhood. The major neurological feature is hypotonia that typically is recognized within a year of life. This evolved into ataxia and developmental delay, often associated with intellectual deficit, abnormal breathing pattern and abnormal eye movements. There may be alternate episodes of apnea and tachypnea, which tend to occur shortly after birth and may improve with age. Recurrent or prolonged apnea may require ventilatory assisted ventilation. There may be nystagmus with strabismus and ptosis. Facial features are broad forehead, arched eyebrows, ptosis, hypertelorism, low- set ears, and a triangle- shaped mouth. Magnetic resonance imaging shows a “molar tooth sign” in which the cerebellar vermis of the brain is absent or underdeveloped and the brain stem is abnormal. Asphyxiating thoracic dystrophy or Jeune syndrome is a rare autosomal recessive ciliopathy characterized by multiple skeleto-muscular abnormalities, multi-organ involvement and variable severity [16]. Mutations in at least 11 genes have been found. Mutations in gene, DYNC2H1, accounts for up to half of all cases. The newborn presents with a pulmonary hypoplasia with shortened ribs and short limbs. Subsequently these patients develop renal dysfunction, hepatic disorder, and retinal dystrophy. Some have growth delay, GI abnormalities, recurrent rectal prolapse, and congestive cardiac failure. There may be associated hematuria, proteinuria, and inability to concentrate the urine. Serious thoracic dystrophy may lead to hypoxemia and respiratory failure. There may be a need for an early tracheostomy. Prenatal studies may show polyhydramnios and narrow thorax, and short bones. Supportive therapy is required for management of respiratory compromise.
Sensenbrenner syndrome or cranioectodermal dysplasia is a ciliopathy that presents with craniofacial, skeletal, and ectodermal abnormalities. They have short stature, limb shortening, short ribs, narrow chest, brachydactyly, renal failure, and hepatic fibrosis. It has been related to homozygous missense mutation in the IFT122 (WDR10) gene [17]. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene [17].
Senior-Loken syndrome is an autosomal recessive ciliopathy. Mutations in 10 different genes, NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, SDCCAG8, WDR19, CEP164 and TRAF3IP1, have been reported [18]. Renal cysts develop early in childhood, they present with polyuria, polydipsia, renal dysfunction, generalized diffuse hypotonia and fatigue. End-stage renal disease develops in childhood or in adolescence. This condition causes an increased sensitivity to light, nystagmus, and refractory problems. There is risk for Leber congenital amaurosis within the first few years of life or in childhood.
Boucher-Neuhauser Syndrome is a condition that results in ataxic movement abnormalities, hypogonadotropic hypogonadism and choroidal-retinal degeneration, resulting in vision loss [19]. They may sometimes demonstrate developmental delays or rapid side-to-side movement of the eyes called nystagmus. This condition has been associated with mutations in the PNPLA6 gene.
Gordon-Holmes Syndrome presents with ataxia and hypogonadism. This condition is associated with mutations in the PNPLA6 gene. This is an autosomal recessive, usually adult-onset neurodegenerative disease characterized by cognitive decline and dementia [20].
Oliver-McFarlane Syndrome is an autosomal recessive disorder that presents with intrauterine growth retardation, choroid and retina dystrophy, intellectual disability, and hypopituitarism. Patients have facial dysmorphism presenting with a large frontal skull, prominent chin, mid- front scalp hair loss, bushy eyebrows, and long eyelashes [21]. This condition is associated with mutations in the PNPLA6. Initial peripheral visual field leads first to night blindness and eventually to total blindness.
Clinical Management of Bardet Biedl Syndrome
These infants require multidisciplinary follow-up. The primary care provider needs to have a follow-up with geneticist, nephrologist, ophthalmologist, cardiologist, neurologist, behavioral medicine experts, audiologist, physical therapist, dietitian, and endocrinologist. Blood pressure should be measured six monthly or more often if there is evidence of hypertension. Antihypertensives and lipid-lowering medication should be prescribed as appropriate. It is recommended that all the patients have at least one baseline renal ultrasound to rule out any obvious malformations. Patients with renal impairment should be referred to a nephrologist. A detailed ophthalmological assessment including an electroretinogram is required to determine the onset and degree of rod-cone dystrophy and to screen for refractive errors, retinopathy, or cataracts. Visual aids and mobility training can enhance physical activity. Obesity and metabolic syndromes require careful weight control. Carefully crafted exercise therapy along with diet management is crucial for weight control.
The management of patients with BBS is geared to management of specific presentations. They are enrolled in early intervention program with multi-disciplinary follow-up with endocrinologist, ophthalmologist, geneticist, nephrologist, audiologist, cardiologist, physical therapist, dietitian, and primary care provider.
Endocrinopathies related to lower hormones may require supplementation under the support of endocrinologist. Endocrinological assessment is important because the risk of diabetes mellitus, thyroid dysfunction, metabolic syndromes, and possible delay in sexual maturity. Endocrinology workup including pituitary function testing may necessitate hormone replacement therapy. They may require specialized education with dietary control and exercise based on their abilities. Long- term follow-up may necessitate bariatric surgery. Vitamin and mineral supplementation may support best function of visual and auditory function. Regular eye exam is required for management of refractory errors. Electroretinogram is an important tool for ophthalmological assessment. The patients need to be followed up for developmental and educational assessment. Early intervention along with physical therapy, occupational therapy and speech therapy is optimum for the best educational environment. Follow-
up by clinical psychologist is important to deal with minor or major behavioural concerns. Orthodontist assessment may be required for high-arched palate, dental crowding, and abnormal teething. A follow-up by cardiologist as recommended for structural cardiac abnormalities. Genetic counselling is recommended for the individual and the family members. The family of our index patient has been offered genetic testing.
Currently, there are some specialized management plans with potential use of stem cells, to be reprogrammed and differentiated into different cells to manage ciliopathy- related eye disease and nephropathy. Retinal gene therapy has been successful with Leber congenital amaurosis. Exon skipping therapy targets the mutation and has been found to be useful in clinical trials of Duchenne muscular dystrophy and spinal muscular atrophy. There are some futures goals for genomic editing that may allow DNA to be replaced, corrected, or removed.
Genetic counselling is important tool in this disorder. It is therefore appropriate to counsel patients and families according for a typical autosomal recessive recurrence risk. Sibling of an affected child has a 25% risk of being affected, 50% risk of being an asymptomatic carrier and 25% of being unaffected and not a carrier. If there is a known mutation in the family, prenatal testing should be started early. Prenatal sonography may be helpful if there is an associated polydactyly, renal, cardiac, or other association. There is a high risk of infertility.
References
-
Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, et al. (1989) The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 321(15): 1002-1009.
-
Mhamdi O, Ouertani I, Bouhamed HC (2014) Update on the genetics of bardet-biedl syndrome. Mol Syndromol 5(2): 51-56.
-
Forsyth RL, Aygun GM (2003) Bardet-Biedl Syndrome Overview. In: Adam MP, et al. (Edn.), GeneReviews® [Internet]. Seattle (WA): University of Washington, USA.
-
Forsythe E, Beales PL (2013) Bardet-Biedl syndrome. Eur J Hum Genet 21(1): 8-13.
-
Hamel CP (2007) Cone rod dystrophies. Orphanet J Rare Dis 2: 7.
-
Baker K, Beales PL (2009) Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 151C (4): 281-295.
-
Putoux A, Bitach TA, Martinovic J, Gubler MC (2012) Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol 27(1): 7-15.
-
Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, et al. (1996) The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis 27(6): 776-783.
-
Slavotinek AM (2002) McKusick-Kaufman Syndrome. 2002. In: Adam MP, et al. (Edn.), GeneReviews®, University of Washington, Seattle, USA.
-
Kumar A, Husain AS, Saleem A, Khawaja UA, Virani S (2020) Laurence-Moon-Bardet-Biedl Syndrome: A Rare Case With a Literature Review. Cureus 12(11): e11355.
-
Joy T, Cao H, Black G, Malik R, Menys VC, et al. (2007) Alstrom syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis 2: 49.
-
Parelkar SV, Kapadnis SP, Sanghvi BV, Joshi PB, Mundada D, et al. (2013) Meckel-Gruber syndrome: A rare and lethal anomaly with review of literature. J Pediatr Neurosci 8(2): 154-157.
-
Verloes A, Temple IK, Bonnet S, Bottani A (1997) Coloboma, mental retardation, hypogonadism, and obesity: critical review of the so-called Biemond syndrome type 2, updated nosology, and delineation of three “new” syndromes. Am J Med Genet 69(4): 370-379.
-
Driscoll DJ, Miller JL, Schwartz S, Cassidy SB (1993) Prader-Willi Syndrome. In: Adam MP, et al. (Edn.), GeneReviews®, University of Washington, Seattle, USA.
-
Brancati F, Dallapiccola B, Valente EM (2010) Joubert Syndrome and related disorders. Orphanet J Rare Dis 5: 20.
-
Poyner SE, Bradshaw WT (2013) Jeune syndrome: considerations for management of asphyxiating thoracic dystrophy. Neonatal Netw 32(5): 342-52.
-
Sztulpa JW, Eggenschwiler J, Osborn D, Brown DA, Emma F, et al. (2010) Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 86(6): 949-956.
-
Caridi G, Murer L, Bellantuono R, Sorino P, Caringella DA, et al. (1998) Renal- retinal syndromes: association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am J Kidney Dis 32(6): 1059-1062.
-
Neuhauser G, Opitz JM (1975) Autosomal recessive syndrome of cerebellar ataxia and hypogonadotropic hypogonadism. Clin Genet 7(5): 426-434.
-
Calandra CR, Mocarbel Y, Vishnopolska SA, Toneguzzo V, Oliveri J, et al. (2019) Gordon Holmes Syndrome Caused by RNF216 Novel Mutation in 2 Argentinean Siblings. Mov Disord Clin Pract 6(3): 259-262.
-
Haimi M (2005) Gershoni-Baruch, R. Am J Med Genet 138A: 268-271.
- Understanding Pediatric Multiple Sclerosis: Clinical Presentation, Diagnostic Criteria, Therapeutic Advances, and Supportive Care Approaches
- Hemophilia in Children
- Xia-Gibbs Syndrome- A Case Report
- A Study to Assess Effectiveness of Play Therapy in Reducing Post-Operative Pain among Children Age 2 To 5 Year who have Undergone General Surgeries in Selected Pediatric Hospitals of Vadodara
- Preterm Birth: Scope of the Problem, Cost of Care, Potential Complications and Current Guidelines for Management
- Noradrenaline: Can we Use it to Manage Hemodynamic Instability among Neonatal Septic Shock at the NICU?