
Citation: Shahin A et al. Study of Genetics Mutations in HEXA Gene for Induced Tay-Sachs Disease in Human, Tabriz, Iran. J Microbiol Biotechnol, 2017, 2(1): 000113.
*Corresponding author: Shahin Asadi, Student of Molecular Genetics, Young Researchers and Elite Club, Tabriz Branch, Islamic Azad University, Tabriz, Iran, Tel: +989379923364 ;Email: hahin.asadi1985@gmail.com
Tay-Sachs disease is one of the few neurodegenerative diseases of known causes. It results from mutations of the HEXA gene encoding the alpha subunit of beta-hexosaminidase, producing a destructive ganglioside accumulation in lysosomes, principally in neurons. With the determination of the protein sequence of the alpha and beta subunits, deduced from cDNA sequences, the complex pathway of sub cellular and lysosomal processing of the enzyme has been determined. More recently, detailed knowledge of the gene structure has allowed the determination of specific mutations causing TaySachs disease. The high incidence of the disease in Ashkenazi Jews is attributed predominantly to three mutations present in high frequency, while in non-Jews some two dozen mutations have been identified thus far. The cataloguing of mutations has important implications for carrier screening and prenatal diagnosis for Tay-Sachs disease
Keywords: HEXA Gene; cDNA; Beta-Hexosaminidase; Tay-Sachs Disease
Tay–Sachs disease (also known as GM2 gangliosidosis or hexosaminidase a deficiency) is a rare autosomal recessive genetic disease. In its most common allele, it causes a progressive deterioration of nerve cells and of mental and physical abilities that begins around 7 months of age and usually results in death by the age of four. The disease occurs when harmful quantities of cell membrane components known as gangliosides accumulate in the brain's nerve cells, eventually leading to the premature death of the cells. A ganglioside is a form of sphingolipid, which makes Tay–Sachs disease a member of the sphingolipidoses. There is no known cure or treatment [1- 19].
Material and MethodsIn this study, 130 patients with Tay-Sachs disease and 300 healthy controls were studied. Peripheral blood samples from patients and parents with written permission control were prepared. After separation of serum, using Real Time-PCR technique of RNA molecules was collected. To isolate Neuroglial cells erythrocytes were precipitated from hydroxyethyl starch (HES) was used. At this stage, HES solution in ratio of 1 to 5 with the peripheral blood of patients and controls were mixed. After 60 minutes of incubation at room temperature, the supernatant was removed and centrifuged for 9 min at 200 Gera [20]. The cell sediment with PBS (phosphate buffered saline), pipetazh and slowly soluble carbohydrate ratio of 1to2 on ficole (Ficoll) was poured in the 500G was centrifuged for 24 minutes. Mono nuclear Neuroglial cells also are included, has a lower density than ficole and soon which they are based. The remaining erythrocytes have a molecular weight greater than ficole and deposited in test tubes [21-24].
The supernatant, which contained the mono nuclear cells, was removed, and the 300 G was centrifuged for 12 minutes. Finally, the sediment cell, the antibody and Neuroglial cells was added after 34 minutes incubation at 5° C, the cell mixture was passed from pillar HEXA. Then the cells were washed with PBS and attached to the column HEXA pam Stem cell culture medium containing the transcription gene HEXA and were kept [23].
To determine the purity of Neuroglial cells are extracted, flow cytometry was used. For this purpose, approximately 8-9 × 104 Neuroglial cells were transfer red to1.5ml Eppendorf tube and then was centrifuged at 2000 rpm for 7minutes at time. Remove the supernatant culture medium and there maining sediment, 100μl of PBS buffer was added. After adding 5-10μl CD38+ PE monoclonal anti body to the cell suspension for 60 min at 4°C, incubated and read immediately by flow cytometry Technique. For example, rather than control anti body Neuroglial cells PE, IgG1 negative control solution was used [22].
Total RNA Extraction Procedure Includes2ml solution spilled Qiazolon cells, and slowly and carefully mixed and incubated at room temperature for 5 minutes. Then 100μl chloroform solution to target mix, then transfer the micro tubes were added, and the shaker well was mixed for 15 seconds. The present mix for 4 minutes at room temperature and then incubated for 20 min at 5°C an was centrifuged at 13200 rpm era. Remove the upper phase products were transferred to a new microtube and to the one times the volume of cold ethanol were added. The resulting mixtures for 24 hours at -20°C were incubated [25].
Then for 55min at 5°C was centrifuged at 12800 rpm era. Remove the supernatant and the white precipitate, 1ml of cold 75% ethanol was added to separate the sediment from micro tubes were vortex well. The resulting mixture for 20 min at 5°C by the time we were centrifuged 12800 rpm. Ethanol and the sediment was removed and placed at room temperature until completely dry deposition. The precipitate was dissolved in 20μl sterile water and at a later stage, the concentration of extracted RNA was determined [26-30].
Discussion and ConclusionAccording to the results of sequencing the genome of patients with Tay-Sachs disease, and the genetic mutations HEXA gene found that about 100% of patients with Tay-Sachs disease, they have these genetic mutations. Patients with Tay-Sachs disease, unusual and frightening images in the process of Tay-Sachs disease, experience. Lot epigenetic factors involved in Tay-Sachs disease. But the most prominent factor to induce TaySachs disease, mutations is HEXA gene. This gene can induce the birth and can also be induced in the adulthood.
AcknowledgmentsThanks to everyone who helped us in doing this project, very grateful. Patients and their families also accept patients who had very much for your cooperation in this study.
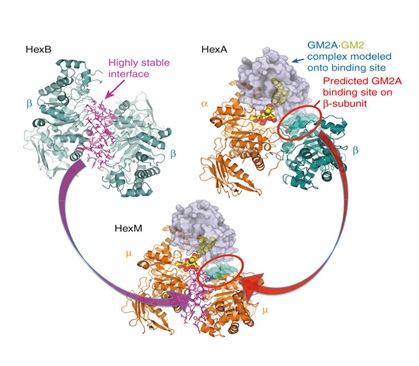
Figure 1: Schematic view of protein structure HEXA and HEXB and GM2 in human.
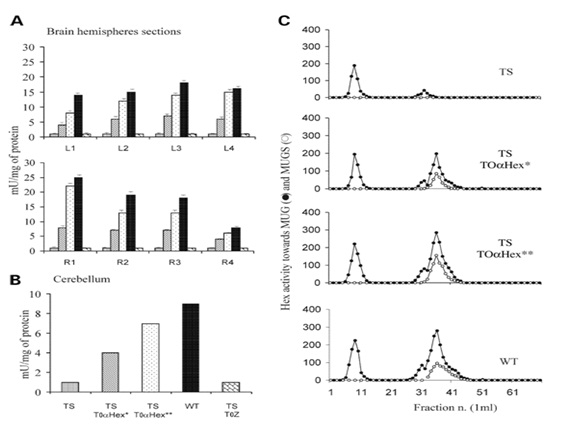
Figure 2: Schematic view of the HEX protein expression in brain cells of patients.
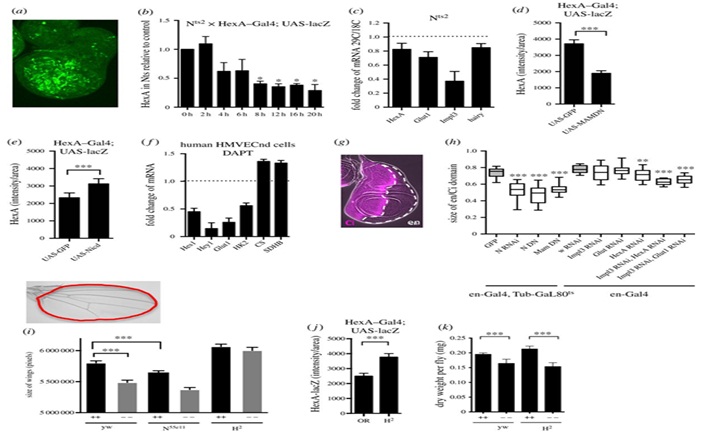
Figure 3: Schematic view of linear and microscopic HEXA protein expression level in the brain cells of patients.
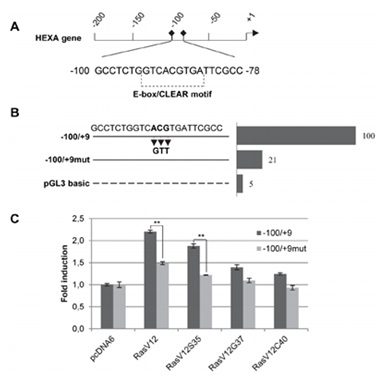
Figure 4: Schematic view of the mutation HEXA gene sequence.
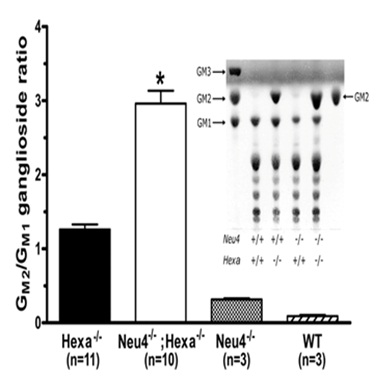
Figure 5: Schematic view of the pattern formed band HEXA gene.
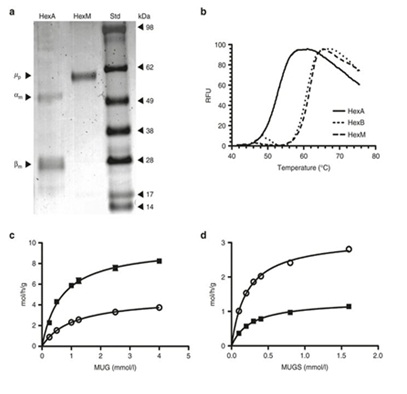
Figure 6: Schematic view of denaturing HEXA gene.
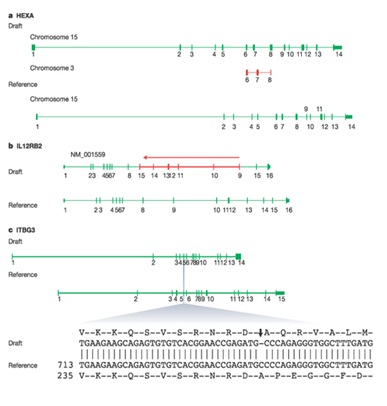
Figure 7: Schematic view of the HEXA gene sequence of amino acids in codon.
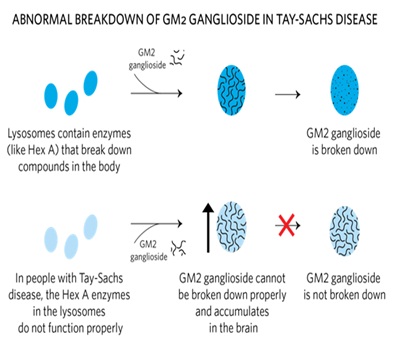
Figure 8: Schematic view of abnormal breakdown of GM2 ganglioside in Tay-Sachs disease.




