
Citation: Noor HN. β-Carbonic Anhydrase as a Target for Eradication of Mycobacterium tuberculosis. J Pharm Res 2017, 1(1): 000106.
*Corresponding author: Noor H Naser, PhD, Department of Pharmaceutical Chemistry, College of Pharmacy, University of Kufa, Najaf, Iraq Email: noorh.naser@uokufa.edu.iq
Infectious diseases are among the most danger, and deadly afflictions that human societies suffering from there. One of the common re-emerged infectious diseases is tuberculosis (TB). The causative pathogen for this disease has developed several methods to resist the ordinary therapy, which are usually acting through inhibition of nucleic acids. Therefore, new researches focused on development of tuberculosis treatment, through discovery of novel targets, on which new synthesized drugs will act. One of these targets that can be utilized for eradication of tuberculosis is carbonic anhydrase enzyme.
Keywords: β-Carbonic Anhydrase; Mycobacterium tuberculosis; Infectious diseases
Tuberculosis is one of the ancient infectious disease which has been re-emerged, long after the medical establishment was considered it to be declined [1]. The re-emergence is particularly due to the development of resistance to the ordinary therapy [2]. The appearance of multi-drug resistance tuberculosis (MDR-TB), and extensively drug resistance tuberculosis (XDR-TB), made a great obstacle for controlling of this infectious disease [3].Tuberculosis is a global airborne disease, which caused by gram positive bacterium Mycobacterium tuberculosis (Mtb) [4]. This bacteria is non-motile, nonspore forming, aerobic rods, its cell wall is highly hydrophobic, due to richness with different types of lipids [5]. The ordinary anti-tuberculosis drugs can be classified into; First line drugs; include Isoniazide (INH), Rifampicin, Ethambutol, Pyrazinamide, and Streptomycin [6]. Second line drugs; include Amikacin, Capreomycin, Cycloserine, Thiacetazone, Ethionamide, Para-aminosalicylic acid, and quinolones as ciprofloxacin.
MDR-TB, mean the simultaneous resistance to one or more of the five first line drugs [3], while the XDR-TB, mean the resistance to at least Isoniazid and Rifampicin in addition to one of quinolones, and at least one of the injectable second line drugs as Amikacin [7].
The resistance situation demonstrates a demand for development of novel approaches for TB treatment. A potential method to overcome the resistance problem is to design, and synthesize novel and innovative chemical agents that act on novel targets, through different mechanisms of action.
Carbonic Anhydrase as New Target for Tb TreatmentCarbonic an hydrase is a metallo enzyme, which catalyze the hydration of carbon dioxide to bicarbonate, by that control the pH homeostasis inside the cell [8]. This enzyme is present in both eukaryotes, and prokaryotes, and encoded by five gene families; α (in vertebrates), β (in bacteria and fungi), γ (archaea), δ and ζ(in marine diatoms) [9]. Mycobacterium tuberculosis contains three β-carbonic anhydrase (CA) genes in its genome, that encoding for three proteins, named mtCA 1, mtCA 2 and a third enzyme, mtCA 3 [10, 11].
Catalytic Mechanism of Carbonic AnhydraseIn all enzyme classes, a metal hydroxide species (L3–
M2+–OH) of the enzyme is the catalytically active species,
acting as a strong nucleophile (at neutral pH) on the CO
The second stage is the transfer of a proton from the zinc bound water to bulk solvent to regenerate the zincbound hydroxide. Here B is a proton acceptor in solution or a residue of the enzyme itself [12].
In many enzymes, generation of the metal hydroxide species from the metal-coordinated water is the rate determining step of the catalytic turnover, making CAs among the most effective catalysts known in nature.
The metal ion ligands are three His residues in α -, γ -, and δ -CAs or one His and two Cys residues in β - and ζ – CAs [13]. Some β-class enzymes have four protein zinc ligands, that is, one His, two Cys, and one Asp coordinated to Zn (II). This coordination setup (denoted "blocked") leaves no room for a water or hydroxide ligand and seems incompatible with a zinc-hydroxide mechanism. This apparent inconsistency has led to alternative mechanistic proposals, and also the suggestion that the aspartate may be involved in activity regulation of the enzyme by somehow changing between the accessible and blocked states. For these enzymes no water coordinated to the metal ion is present at pH values <8. However, at pH values >8, a conserved Arg residue in all β-CAs investigated so far (belonging to a so called catalytic dyad), makes a salt bridge with the Asp coordinated to Zn (II), liberating the fourth Zn (II) coordination position, which is then occupied by an incoming water molecule/hydroxide ion, as shown in the structures of two β-CAs from Mycobacterium tuberculosis, encoded by Rv1284 and Rv3588c genes [10].
The cavity is partitioned into two very different environments. On one side of the zinc, deep within the active site, lies a cluster of hydrophobic amino acids (namely Val, Leu, Thr, and Trp.), whereas on the other side of the zinc, leading out of the active site to the bulk solvent, the surface is lined with hydrophilic amino acids (namely Tyr, Asn, His, Thr-199-O_1, and Thr-200-O_1) [8, 12].
Carbonic Anhydrase InhibitorsThere are three main classes of inhibitors [14].
These inhibitors bind to the Zn2+ ion of the enzyme either by substituting the non-protein zinc ligand to generate a tetrahedral adduct, or by addition to the metal coordination sphere to generate trigonal-bipyramidal species [15]. As shown in the following equations [8];

The unsubstituted sulfonamides and their bioisosteres, which are the most important CAIs, bind in the tetrahedral geometry of the Zn2+ ion of the enzyme (Figure 3A), in deprotonated state, with the nitrogen atom of the sulfonamide moiety coordinated to Zn2+ and an extended network of hydrogen bonds involving residues Thr and Glu, also participating to the anchoring of the inhibitor molecule to the metal ion. While the anions add to the metal coordination sphere, generating trigonalbipyramidal specie, such as the thiocyanate adduct shown in (Figure 3B) [16].
The aromatic/heteroaromatic part of the inhibitor (R) viz; sulfonamide/sulfamate/sulfamide interact with the hydrophilic and hydrophobic residues of carbonic anhydrase. In deprotonated state as SO2NH it is coordinated to Zn+2 of the enzyme, and it is –NH- moiety participate in the hydrogen bond with the oxygen of Thr., which in turn is engaged in another hydrogen bond to the carboxylate group of Glu. One of the oxygen atoms of the – SO2NH- moiety also participates in a hydrogen bond with the backbone –N- moiety of Thr [17].
A general pharmacophore (Figure 4), for the compounds acting as carbonic anhydrase inhibitors has been reported by Thiry et al. from the analysis of the CA active site and from the structure of the inhibitors described in the literature. This pharmacophore includes the structural elements that are required to be present in the compounds in order to act as CA inhibitors. This includes the presence of a sulfonamide moiety which coordinates with the zinc ion of the active site of the CA and the sulfonamide is attached to a scaffold which is usually a benzene ring. The side chain might possess a hydrophilic link able to interact with the hydrophilic part of the active site and a hydrophobic moiety which can interact with the hydrophobic part of the CA active site.
It was determined that is the nature of the R group that greatly influences the binding to the enzyme. Thus, the nature of the R moiety is the determining factor in controlling the inhibitory power, probably due to flexibility of the linker and the possibility of the R group to orientate in different sub-pockets of the active site cavity of these enzymes. It may be observed that the elongation of the inhibitor molecule, by means of a sulfanilyl moiety, such as in compounds 1, 2, and 3 leads to a roughly 10 times increase of the inhibitory power of the corresponding sulfonamide against mtCA 2, which may be an important hint for drug design purposes [18,19].
 Conclusion
Conclusion
The development of resistance against most of the ordinary therapeutic agents of infectious diseases represents a pressing problem, and required the development of the clinical therapeutic compounds to be effective against previously unexploited targets. Many carbonic anhydrases isolated from organisms open a new therapeutic target, such as α-CA from Plasmodium falciparum and Helicobacter pylori, and β-CA from Mycobacterium tuberculosis and Candida albicans etc. Researchers are being carried out for developing specific inhibitors targeting these enzymes that would lead to conceptually novel therapies.
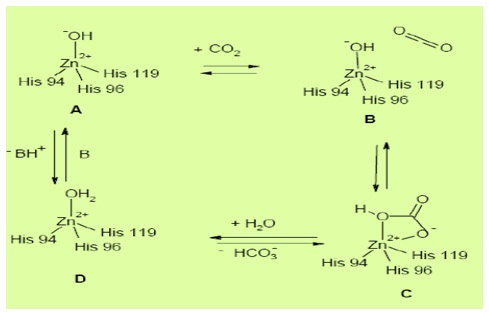
Figure 1: Active site and mechanism of action of carbonic anhydrases [8].
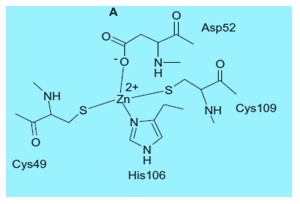
Figure 2: β-carbonic anhydrase active site obtained from Mycobacterium tuberculosis [13].
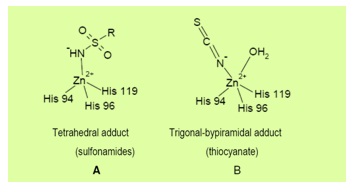
Figure 3: Binding of inhibitors with carbonic anhydrase active site [8].
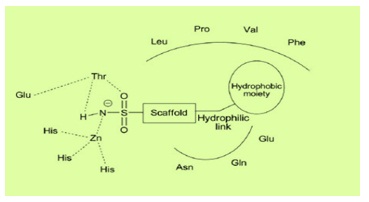
Figure 4: Structural elements of CA inhibitors in the CA enzymatic active site [8].
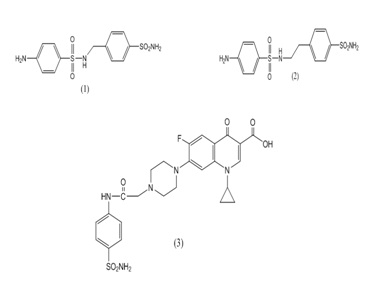
Figure 5




