ASNP: A Personalized Alternative Splicing Neoantigen Discovery Pipeline
Neoantigen is important for immunotherapy. At present, neoantigen prediction is designed mainly based on gene mutations, but there are no methods to predict neoantigens from alternative splicing. We developed a novel ASNP (a personalized alternative splicing neoantigen discovery) pipeline based on alternative splicing information. ASNP uses a new selection strategy to capture potential neoantigen candidates from short peptides generated by alternative splicing, and perform HLA binding affinity prediction to identify high-affinity neoantigen candidates. Then, we applied the pipeline to non-small cell carcinoma and found 21302 potential HLA-peptide pairs. Finally, we evaluated our prediction results through the IEDB database and found 4 experimentally confirmed epitopes among potential candidates. The application of ASNP can help find potential neoantigen candidates and quickly develop personalized immunotherapy for cancer patients.
Introduction
Neoantigen is a peptide that exists only in cancer cells, and it is an epitope-specific antigen produced by tumor cell mutation. Since neoantigens are only expressed in tumor cells rather than normal cells, they are ideal targets for immune cells to attack without harming normal cells. Therefore, targeting neoantigens is an effective way to individualize tumor treatment. The high mutation rate of cancers such as melanoma may carry more neoantigens, and the higher the number of neoantigens, the easier it is for the immune system to track them. This is why neoantigen vaccines are easy to be applied in tumors with high mutation burdens, but difficult to apply in other tumors. In addition to gene mutations, gene fusion and alternative splicing can also generate tumor-specific genes, thereby enriching the number of neoantigens. Such tumor-specific genes can play important roles in neoantigen discovery of the cancers with low mutation burden. To date, mutation-based neoantigen discovery workflow like pVAC-Seq [1], gene-fusion based pipeline like INTERGRATE-Neo [2], and integration pipeline like TSNAD [3] have been constructed, but alternative splicing-based neoantigen discovery pipeline has not been constructed. In this study, we propose a new pipeline, ASNP (Alternative Splicing Neoantigen Pipeline) to identify neoantigens from alternative splicing data of tumor-normal pairs. We select samples from the cancer genome map and apply ASNP to predict neoantigens. By predicting and verifying the results, we showed that ASNP could effectively discover potential new neoantigens.
Materials and Methods
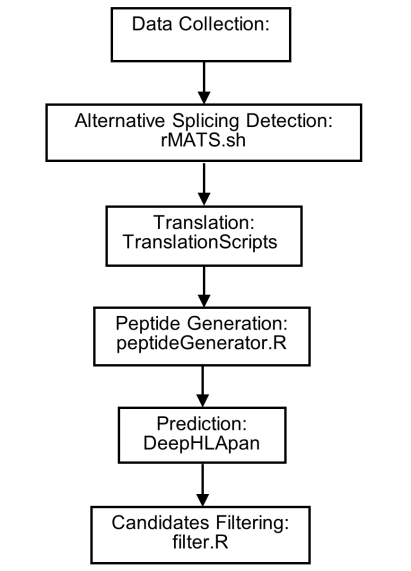
Pipeline Overview
The working flowchart of ASNP is shown in Figure 1. First, patient samples are collected and sequenced to obtain the mRNA data. Next, using alternative splicing detection approach, we can identify high-confidence and tumor-related mRNA sequences, which can be translated into proteins. Because short peptides with a length of 8 to 14 can bind to HLA, and only short peptides composed of amino acids from two genes contain specific information, we therefore set some constraints to generate candidate short peptides. Finally, we used DeepHLApan method to predict the HLA-peptide pairs affinity, and we obtained the peptides as potential neoantigens.
Data
This study used the RNA-Seq data set (GSE37765) [4]. A total of 12 samples were included, containing primary lung tumors and matched normal tissues isolated from 6 Korean female non-small cell lung cancer (NSCLC) patients. The RNA-Seq data set was sequenced using the Solexa sequencing technology platform (Genome Analyzer IIx, Illumina, San Diego, CA). The original data of GSE37765 was downloaded from the European Nucleotide Archive (ENA). FastQC [5] and Trimmomatic [6] were conducted for the quality control process, including adaptor pruning, low-quality bases and short read removal of raw data.
Alternative Splicing Detection
Alternative splicing (AS) is an important mechanism of eukaryotic gene regulation. Splicing is an important step in the processing of multi-exon gene RNA, in which introns are removed from the precursor RNA to produce mature RNA containing splice junctions. In this work, in order to identify AS events related to tumor patients, rMATS [7] with default parameters was used to screen the difference AS in tumor- normal paired samples. GRCh38 (v33) is used as a reference resource genome library downloaded from the CTAT resource genome library website. Finally, five main types of alternative splicing were screened out, including skipping exons (SE), reserved introns (RI), alternative 5’splice sites (A5SS), alternative 3’splice sites (A3SS), and mutually exclusive exons (MXE). The AS satisfied the criteria of FDR ≤ 0.05 and the absolute value of delta PSI = 1 was identified as tumor-specific differential alternative splicing (DAS) genes between tumor sample and normal paired sample.
Translation
For translation, the BED Tools suite [8] was used to extract the nucleotide transcript sequences of tumor-specific DAS genes from human reference genome GRCh38. Then, python scripts were used to translate nucleotide sequences into peptide sequences according to the correspondence between codons and amino acids. The translation started with the first start codon (AUG) of 5’ end and ended with the appearance of the first stop codon in a transcript. Finally, we extracted peptides with 26 amino acids in length that contain 13 amino acids at both sides of the tumor-specific junction point for the next step of analysis.
Peptide Generation
Since only a part of the sub-peptides of a peptide obtained from an AS are potential neoantigen candidates, predicting all sub-peptides is not a good idea. We designed a new method to search for possible peptide candidates from an AS peptide based on two constraints.
The first constraint is that the peptide candidate must contain a junction. Assume there are consecutive genes A, B, and C in cancer patients, and gene B is lost due to cancer. So the junction of A and C contains cancer-specific information and there may be potential neoantigens. Therefore, when searching peptides, we need to find short peptides that contain amino acids produced by two genes at the same time.
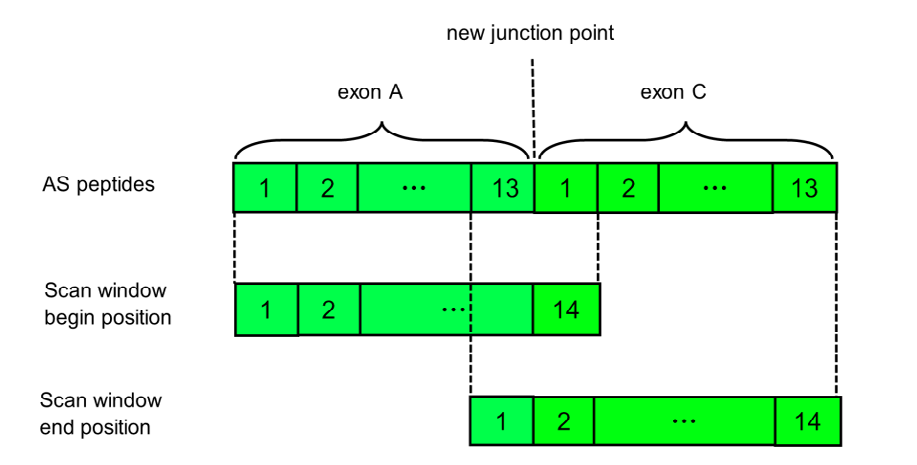
The second constraint is that the length of the peptide candidates is 8 to 14. Most of the peptides with high HLA binding affinity are between 8 and 14 [9], so we took 13 amino acids on each side of the junction of A and C to form an alternative splicing short peptide with a length of 26. In Figure 2, we show how to use the scan window to generate neoantigens. The translated peptide length of the alternative splicing gene was fixed at 26, including 13 amino acids for the A gene and 13 amino acids for the C gene. We used a window size of 8 to 14 to scan the peptides from left to right, while ensuring that the window contained at least one amino acid and another gene. The window snapshot was a possible peptide. Through this step, we generated peptide candidates for the neoantigen prediction.
Prediction
We chose the DeepHLApan (v1.1.1) [10] modesl to predict the binding affinity and immunogenicity of HLA-peptide pairs. DeepHLApan consists of a combination model and an immunogenicity model. For combination model, 437,077 HLA-pairs were collected as the training data, of which 280,525 were from Immune Epitope Database (IEDB) and 156,552 were pseudo-pairs. Similarly for immunogenicity model, 32,785 immunogenicity data were collected from IEDB. The training model architecture uses the bidirectional gated recurrent unit (BiGRU) tree layer and attention layer to improve prediction accuracy. DeepHLApan can accurately predict the binding score and immunogenicity score, which can help us locate the true neoantigen.
Results
Our objective is to develop an alternative splicing pipeline and apply this pipeline to construct a non-small cell carcinoma neoantigen candidate library. First, we selected RNA sequence data from 12 samples of 6 patients. Each patient must have a tumor-normal sample pair for differential alternative splicing analysis. These data were processed by rMATS to identify alternative splicing events, and we obtained short peptides of 26 amino acids. Next, we used these peptides to generate peptide candidates. Each peptide with a length of 8 to 14 and amino acids from different genes are considered as potential peptide candidates. Peptide candidates have different binding affinities to different HLAs, and each patient’s HLA is also different. The higher the affinity between neoantigen and HLA, the stronger the effect of neoantigen vaccine. We hoped to establish a potential library of neonatal antigens, so we need to choose HLAs that appear more frequently in the human body to facilitate the development of personalized treatment plans for different patients. NetMHCpan-4.1 [11] provides information about MHC families, and we selected 12 HLAs (S1 Table) in the ‘HLA supertype representative’ family. By combining candidate peptides with each HLA, we obtained 744,192 HLA-peptide pairs. After removing duplicates, we got 256,428 valid pairs.
To predict binding and immunogenic affinity, we used DeepHLApan to predict these HLA-peptide pairs. For each HLA-peptide pair, a binding score and immunogenicity score were predicted. We selected those pairs with binding score and immunogenicity score greater than 0.9. In the end, we obtained 21, 302 HLA-peptide pairs as neoantigen candidates.
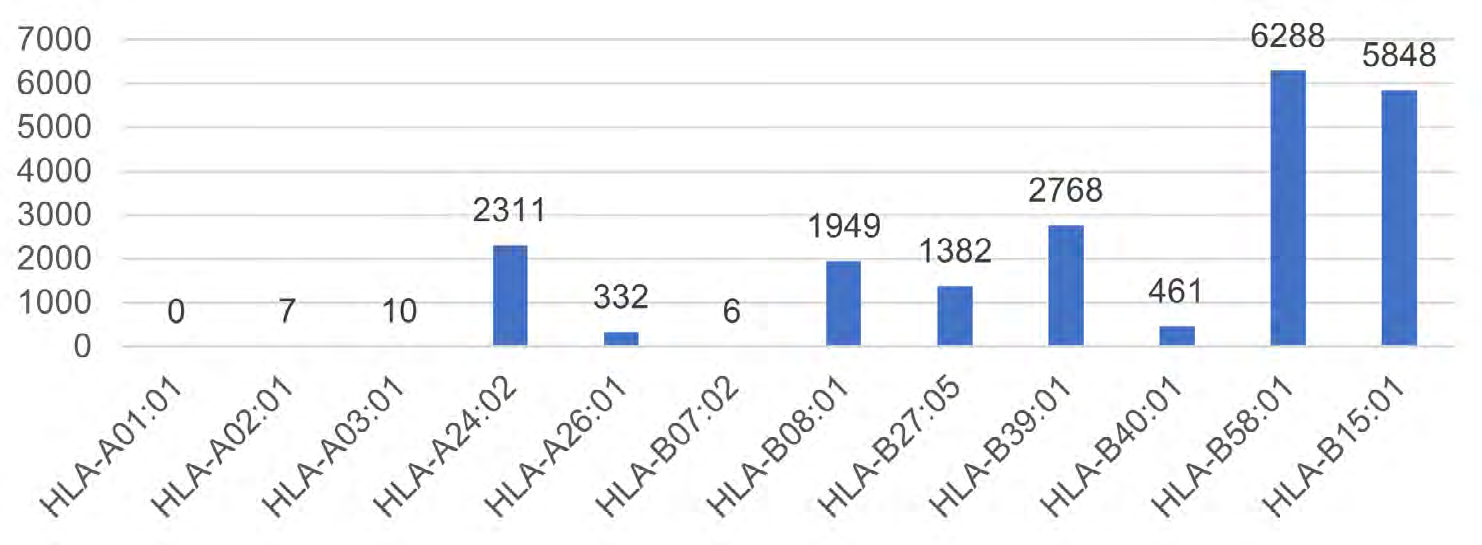
Figure 3 shows the number of predicted neoantigens classified by each HLA type. We found that the neoantigens were concentrated in several HLA types such as HLA-B58:01 and HLA-B15:01. It indicates that lung cancer patients with these HLAs may obtain better therapeutic effects through neoantigen vaccine treatment.
To validate the prediction results, we used the freely available IEDB [12] database. The IEDB catalogs experimental data on antibodies and T cell epitopes studied in humans and other animal species in the context of infectious diseases, allergies, autoimmunity, and transplantation. It contains more than one million epitope data proven by experimental studies. By removing duplicate peptides from 21302 HLA- peptide pairs, we obtained 10094 candidate neoantigens. We searched them from the IEDB database and found 4 epitopes (IEDB epitope ID - 1150525, 1150526, 437988, 1053418, see details in Table 1). This proves that the prediction result of ASNP contains experimentally verified neoantigens, and other unverified candidates are worthy of further mining.
| Epitope ID | Epitope | HLA | Binding score | Immunogenic score | Antigen |
|---|---|---|---|---|---|
| 1150525 | KAGKTLQIFNIEM | HLA-B58:01 | 0.9946 | 0.9757 | Clathrin heavy chain 2 |
| 1150525 | KAGKTLQIFNIEM | HLA-B15:01 | 0.9982 | 0.9346 | Clathrin heavy chain 2 |
| 1150526 | KAGKTLQIFNIEMK | HLA-B58:01 | 0.9715 | 0.9837 | Clathrin heavy chain 2 |
| 1150526 | KAGKTLQIFNIEMK | HLA-B15:01 | 0.9901 | 0.9489 | Clathrin heavy chain 2 |
| 437988 | HPFHATPNTY | HLA-B15:01 | 0.9147 | 0.9081 | Histone-lysine N-me- hlransferase EZH2 |
| 1053418 | TDPGDTASAEARHI | HLA-B07:02 | 0.9244 | 0.9112 | Zinc finger protein Aiolos |
Table 1: Epitopes validated from IEDB with prediction results. The epitope id can be used by search from IEDB.
To further assess the performance of our prediction pipeline on the binding affinity prediction, we compared our approach with the popular prediction method, NetMHCpan. First, we used NetMHCpan to predict the 21302 HLA-peptide pairs, and only 319 pairs were below the weak binding threshold (%Rank <2) and considered as neoantigen candidates. This is much less than the results of ASNP. Second, NetMHCpan can only found one of four proven epitopes. These mean that ASNP can find more and effective neoantigen candidates.
Discussion
Notably, 2 epitopes (IEDB epitope ID - 1150525, 1150526) in Table 1 are derived from Clathrin heavy chain 2 (CHC2) gene. Clathrin is a central components of the clathrin- mediated endocytosis (CME), which is one of the most common mechanisms that cells employ to absorb nutrients, hormones or proteins from the exterior and involves clathrin- coated vesicle. In addition, CME regulates the protein content of the plasma membrane, monitors external cues from the surrounding environment, modulates signaling pathways and directs protein recycling and degradation [13, 14]. Like other membrane-trafficking genes, Clathrin is developmentally and tissue-specifically regulated by alternative splicing [15, 16, 17], to produce multiple transcripts and thus, several protein isoforms with different features. Therefore, the membrane- trafficking genes may serve as an important source for searching alternative splicing-derived neoantigens.
ASNP has two main advantages over similar work, such as INTEGRATE-Neo. First, we did not directly use the variable splicing results to predict the neoantigen, but designed a peptide generation method. This allows us to find more potential candidates. We applied ASNP to prostate patient data (GSE22260) to predict neoantigens. From 160728 HLA- peptide pairs, we found 13207 of them with binding score and immunogenicity score greater than 0.9, a total of 6340 neoantigen candidates, of which 8 are proven epitopes. These are much more than the 240 epitopes of INTEGRATE- Neo results predicted from TCGA-PRAD. On the other hand, our pipeline considers not only binding affinity, but also immune effect. INTEGRATE-Neo filtered 261 HLA-peptide pairs with binding affinity ≤ 500nM. We predicted these HLA-peptide pairs by using DeepHLApan. For considering only the binding score, 191 (73%) HLA-peptide pairs had a binding score greater than 0.9. However, after considering the immunogenicity score, only 46 (18%) HLA-peptide pairs met the above threshold. This shows that our method can filter out candidates whose immune effect may be poor despite high binding affinity.
The limitation of our pipeline is that ASNP predicts too many candidates to experiment. For this issue, we suggest to set an higher threshold to reduce the candidates number. For instance, in this work, we can selected 111 neoantigen candidates (S2 Table) with binding scores and immune scores greater than 0.99 to test their immune effects by using human peripheral blood.
Conclusion
In this study, we developed a new alternative splicing neoantigen discovery pipeline ASNP, designed a strategy for selecting potential candidate peptides, and validated and compared the performance of our pipeline with existing methods. Our results showed that ASNP can effectively predict potential specific neoantigens. We applied this method to non-small cell carcinoma, obtained a potential neoantigen library, and identified experimentally verified epitopes. Overall, in addition to the existing neoantigen discovery methods based on intentional somatic mutations and fusion genes, ASNP proposes a new method based on alternative splicing, which fills the gap in this field and helps to discover more potential personalized treatment targets.
Funding
This work was supported by 1.3.5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University, Grant Number: 2019HXFH022.
References
-
Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, et al. (2016) pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med 8(1): 11.
-
Zhang J, Mardis ER, Maher CA (2017) INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery. Bioinformatics 33(4): 555-557.
-
Zhou Z, Lyu X, Wu J, Yang X, Wu S, et al. (2017) TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R Soc Open Sci 4(4): 170050.
-
Kim SC, Jung Y, Park J, Cho S, Seo C, et al. (2013) A High-Dimensional, Deep-Sequencing Study of Lung Adenocarcinoma in Female Never-Smokers. Plos One 8(2): e55596.
-
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom.
-
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
-
Shen SH, Park JW, Lu ZX, Lin L, Henry MD, et al. (2014) rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA 111(51): 5593-5601.
-
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
-
Bulik Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, et al. (2019) Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol 37: 55-63.
-
Wu J, Wang W, Zhang J, Zhou B, Zhao W, et al. (2019) DeepHLApan: A Deep Learning Approach for Neoantigen Prediction Considering Both HLA-Peptide Binding and Immunogenicity. Front Immunol 10: 2559.
-
Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M (2020) NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48(1): 449-54.
-
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, et al. (2019) The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47(1): 339-343.
-
McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12(8): 517-533.
-
Blue RE, Curry EG, Engels NM, Lee EY, Giudice J (2018) How alternative splicing affects membrane-trafficking dynamics. J Cell Sci 131(10): 216465.
-
Moulay G, Laine J, Lemaitre M, Nakamori M, Nishino I, et al. (2020) Alternative splicing of clathrin heavy chain contributes to the switch from coated pits to plaques. J Cell Biol 219(9).
-
Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, et al. (2013) mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci 16(4): 499-506.
-
Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, et al. (2014) Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun 5: 3603.
- Origin, Evolution, and Functional Impact of Short Insertion- Deletion Variants in Human Genomes: A Review
- Harnessing Molecular Glues for Next-Generation Vaccine, Cancer and Cardiovascular Disease Drug Development: A Comprehensive Review
- Lateral Cervical Epidermal Inclusion Cyst in a Paediatric Patient: A Rare Case Report
- Malarial Plasmodium Falciparum with Hepatitis B and C Virus Infections among Blood Donors in Ife Central Local Government Area, Ile Ife, Osun State, Nigeria
- Withanolides and Withaferin A- What’s next in Ashwagandha Research
- Designing of Dual Pulse Photoacoustic Tomography for Imaging of Drug-Response and Tumor Growth