Assessment of Tetracyclines Residues Contamination in Chicken Meat Samples of Algiers Slaughterhouses Level
Food contamination with antimicrobial residues may occur, entailing assessment and monitoring of the contamination level in edible tissue. In that way, 151 chicken meat samples were randomly collected from Algiers slaughterhouses. Otherwise, two analytical methods are validated for the simultaneous determination of oxytetracycline, tetracycline, doxycycline and chlortetracycline residues in edible chicken tissues, by liquid chromatography combined with mass spectrometry detection. Beforehand, an extraction step is performed. Indeed, acetonitrile is used as an extraction solvent for the qualitative method, and then the quantitative method is carried out after a preliminary extraction with an acidified EDTA-McIlvaine buffer, followed by a solid-phase extraction cleanup. Validation qualitative method results were satisfactory in terms of specificity (100%), sensivity, detection capability (CCß) and detection limit (LOD). Furthermore, validation results of the quantitative method was pertinent to the EU commission criteria. Thus, determination coefficient (r2) values were between de 0.91 to 0.98%, where the lowest values were observed for doxycycline. Trueness was between 85.5% and 104.8, expect for tetracycline (76.6% at 150μg/kg) and doxycycline (134.7% at 100μg/kg). As for intraday and interday precision, values respectively ranged between 12.5 and 26% and between 14 and 35%, except doxycycline for which precision values were higher. The methods have been successfully used for the identification, confirmation and quantification of tetracyclines in chicken muscle samples. Consequently, 25 samples were suspects in regard tetracyclines residues. The comparison of these samples with the QC at MRL (100μg/kg) revealed five samples needed a quantification. Thus, oxytetracycline, its epimer and doxycycline identified in samples were quantified. Indeed, all samples contained oxytetracycline and its epimer, doxycycline or both showed levels less than the decision limit (CCα).
Introduction
Several families of antibiotics exist such as beta-lactams i.e. pencillins and cephalosporins, tetracyclines, macrolides, lincosamides, and sulphonamides Mungroo NA, et al. [1] for both human and veterian use. Global antibiotic consumption increased by 65% between 2000 and 2015, from 21.1 billion daily doses determined in 2000 to 34.8 billion in 2015 in 76 countries around the world [2]. The family of tetracyclines alone represents 36.5% of the tonnage sales. Critical antibiotics i.e. latest generation cephalosporins and fluoroquinolones represent nearly 1.0% of the tonnage of active ingredient sold. Over the years 2014 and 2015, the average total sales volume is close to 650 tonnes of antibiotics per year [3]. The problem of antibiotic residues, particularly in meats, is widely discussed by both scientific and media communities.
Indeed, this problem could have a health impact involving not only public health but also economics, thus limiting international trade. Therefore, multidrug resistance to antibiotics is a global threat because the powerful antibiotics available in human clinics are becoming extremely rare [4]. Besides, a Canadian study showed a strong relationship between the commensal E. coli from retail chicken and human infections and the pathogen Salmonella enterica serovar Heidelberg. These results exemplify the fast propagation of resistant bacteria [5].
Moreover, despite the ban on the addition of antibiotics to animal feed in Europe since 2006 Goucem R [6], in the United States and Canada, this practice persists [7]. In Algeria, antibiotic supplementation is probable and could even be common, in the absence of any analytical control of animal feed.
For all these reasons, different analytical methods were set over the world to assess the food contamination rates by drugs residues. European Union (EU) [8] for instance, as well as the Algerian regulation, later in 2016 [9], have defined maximum residue limits (MRLs) for residues of veterinary drugs in food, to protect consumers’ health by ensuring food safety. Maximum residue limits (MRLs) are the higher levels of residue concentrations of pharmacologically active substances legally permitted in or on food and feed. Thus, MRLs for muscle, liver and kidney tissues were established at 100_µg/kg, 300 µg/kg and 600µ_g/kg, respectively. Concerning the three tetracyclines (Tetracycline, Oxytetracycline and Chlortetracycline), the marker residue is the sum of parent drug and its 4-epimer, whereas doxycycline is the marker residue [10].
The objective of our study was assessment of tetracyclines residues contasmination in chicken meat samples of Algiers slaughterhouses level. This study was intended as the starting point for further risk assessments of drugs residues contamination of food in Algeria.
Materials and Methods
Chemicals and Reagents
The antimicrobial standards Tetracycline Hydrochloride, Chlortetracycline Hydrochloride, Demeclocycline Hydrochloride, 4-Epioxytetracycline, 4-Epichlortetracycline Hydrochloride, 4-Epitetracycline Hydrochloride were purchased from Merck Sigma-Aldrich ; Doxycycline Hyclate from Mevet and Oxytetracycline Hydrochloride from Fluka. Sulfaphenazole was obtained from Dr Ehrenstorfer GmbH. Standards were provided with their certificate of analysis, and stored according to supplier’s recommendations.
Acetonitrile for high-performance liquid chromatography (HPLC) and liquid chromatography/mass spectrometry (LC/MS), and Methanol were purchased from Panreac Applichem; Ethylenediaminetetraacetic acid disodium dihydrate (Na2EDTA dihydrate) from Merck Sigma-Aldrich and Trichloroacetic acid (TCA) was obtained from Panreacquimica SA. Ammonium acetate, Suprapur formic acid were purchased from Merck. Citric acid and Trifluoroacetic acid (TFA) were purchased from TCI Chemicals; and ammonium formate from VWR Chemicals. Sodium dihydrogen phosphate dihydrate was obtained from AlfaAesar.
Millipore Milli-Q-Plus ultrapure water system was used throughout the study to obtain HPLC grade water used in the preparation of solutions and samples. Oasis HLB extraction cartridges 6 cm3 /200 mg were purchased from Waters.
Solutions Preparation
A solution of sodium dihydrogen phosphate dehydrate at 0.2 mol/L was prepared by dissolving 35.56 g into 1000 mL of water. To obtain a solution of citric acid at 0.1 mol/L, 21g were dissolved into 1000 mL of water.
A solution containing McIlvaine buffer for extraction, was obtained by mixing 1000 mL of citric acid (0.1 mol/L) with 625 mL of disodium hydrogen phosphate (0.2 M). Adjust pH to 4.0 ± 0.05 with NaOH or HCl as needed. Then, Na2EDTA- McIlvaine buffer (0.1 mol/L) was prepared by mixing 60.5g of Na2 EDTA. 2H2O with 1625 mL of McIlvaine buffer.
A solution of trichloroacetic acid 20% was obtained by dissolving 20 g in 100 mL of water.
A rinse solution water/methanol (5%) was prepared by adding 5mL of methanol to 100mL of water.
An elution solution of 1% methanol/TFA (V/V) was prepared by mixing 1mL TFA with 100mL methanol.
A solution of ammonium acetate 2 mol /L (15.4 g in 100 ml of ultrapure water) was prepared and then diluted to 1/10th in ultrapure water, for the final reconstitution of the residue to qualitative method.
A formic acid solution 0.2 mol/L was prepared for the final reconstitution of the residue to confirm and quantify tetracyclines in chicken meat.
Mobile Phase Preparation
Mobile phases A and B for qualitative method were respectively, 0.1% TFA aqueous solution and 0.1% TFA in methanol/acetonitrile (70:30). Mobile phases A and B for quantitative method were respectively, 0.2% ammonium formate with formic acid aqueous solution and 0.2% ammonium formate and formic acid in acetonitrile.
Standard Stock Solutions and Working Solutions
Standard stock solutions 0.5 mg/mL were prepared in methanol independently. The prepared stock solutions were stored below –18°C.
An internal standard stock solution was prepared with ultrapure water to obtain 1 μg/mL of working solution, which was stored at 4°C. Sulfaphenazole was used as an internal standard for the identification of oxytetracycline, tetracycline, chlortetracycline, and doxycycline. Demeclocycline was used for the quantification of the four tetracyclines and theirs epimers. Working solutions were prepared using the stock solution diluted with water. The working solutions were prepared daily. For confirmation and quantification, the working solutions were a mixture of the tetracyclines and their epimers, prepared by serial dilutions of the stock solution in methanol and were stored in brown glass vials at 4°C
Sample Preparation
A 100g sample of chicken meat was homogenized with a Moulinex mixer. Then, 2g were weighted, placed in propylene tubes and stored at 20°C until analysis.
The identification of tetracyclines residues in chicken meat samples was performed by preparing a negative control using a blank sample (free of the interest analytes), and a quality control (QC) which was a fortified sample at a half of the MRL value (50µg/kg) for validation method (MRL i.e 100 µg/kg for samples qualitative analysis purpose). The internal standard used at this stage was sulfaphenazole. A volume of 200 μL of the 1 μg/mL working solution of internal standard was added to all samples before the extraction procedure.
A 2g homogeneous sample (accurate to 0.04 g) was placed into a 50mL polypropylene centrifuge tube with 600µL for fortified samples and 800µL of water for samples to be analyzed and the blank sample. Fortified samples are obtained by adding 200 μL of 0.5 mg/mL tetracyclines working solution.
On the other hand, the confirmation and quantification step was achieved. Indeed, a matrix-matched standard calibration curve was obtained by preparing five tubes, each containing 2±0.04 g of meat chicken. Increasing volumes of the 0.5 mg/mL working solution (10, 20, 30, and 40µL) were added to every tube to obtain 0.5, 1, 1.5, and 2 µg/mL spiking concentrations. Purified water was added to every tube to reach a final volume of 10mL. All samples contained Demeclocycline as an internal standard.
Extraction Procedure
The extraction procedure depends on the type of the analysis method:
- For the qualitative method, tubes containing the prepared samples were kept in the dark for 10 minutes. To 2g amount of homogenized sample, weighed into 15mL polypropylene centrifuge tube, was added 8mL of acetonitrile, which was vortex-mixed for 30sec. After hand, the tubes were shaken 10 min (100 tours/ min) on a rotary shaker. The tubes were centrifuged 5 min at 14000 g at about 4°C, and 6mL of supernatant were decanted carefully into a clean second centrifuge tube. The supernatant was evaporated and dried under nitrogen at 50°C. The residue was dissolved and projected to a constant volume of 0.6mL using the ammonium acetate 0.2mol/L. Then the residue was filtered through a 0.45µm filter membrane and analyzed.
- For the confirmation and the quantification method, tubes containing the prepared samples were kept in the dark for 10 minutes. To 2g amount of homogenized sample, weighed into 50mL polypropylene centrifuge tube, was added 10mL of Na2 EDTA-McIlvaine buffer, which was vortex-mixed for 30sec. The tubes were then stirred for 10 min at 100 revolutionss/min) on a rotary shaker. Subsequently, tubes were centrifuged for 5 min at 14000 g at +4°C. Afterwards, the supernatant was collected in a 50mL polypropylene clean tube. The extraction was repeated with 10mL of Na2 EDTA- McIlvaine buffer. The combined supernatant fluid was mixed with 2mL of trichloracetic acid before freezing for 15 to 20 min. Then, tubes were centrifuged at a rotate speed of 14000g for 10 min at +4°C, and filtered with fast filter paper. The supernatant was purified by using the solid-phase extraction (SPE). The procedure used for the SPE extraction is shown in Figure 1. After evaporation of extracts under nitrogen at 50°C, the dry residue was reconstituted with 500µL of 20% formate acid, filtered through a 0.45µm filter membrane and analyzed.
Liquid Chromatography−Tandem Mass Spectrometry
The analysis was performed on a Perkin Elmer HPLC coupled to an AB Sciex QTrap 3200 triple quadrupole mass spectrometer (MS/MS). All devices being controlled by ANALYST 5.1 software. The analytical column was Symmetry C18, 150 x 3.9mm, 5 μm of particle size from Waters, and heated to 30°C for the tetracyclines identification. Sunfire C18 100x2.1mm column was used at 3°C for the the quantification method. The mobile phase was at a flow rate of 600 μL/min for both methods. The gradient profile for the qualitative method started with 5 min at 81% of eluent A (0.1% TFA aqueous solution) and decreased linearly to 40% at 19min. This composition was increased to 81% at 20min and mobile phase A was then maintained within 5min in the initial conditions. The resulting total run time was 25 min. The mobile phase B consisted of 0.1% TFA in methanol/ acetonitrile (70:30).
The gradient used for the quantitative analysis started with 2 min at 90% of eluent A (0.2% ammonium formate with formic acid aqueous solution) and decreased linearly to 40% within 7 min. This composition was maintained for 2 min and mobile phase A was then increased linearly to 90% at 13 min. With a final equilibration time of 4 min in the initial conditions, the resulting total run time was 17min. The Eluent B contained 0.2% ammonium formate and formic acid in acetonitrile.
| Compound | Precursor ion | Product ion | Time (mSec) | DP | CE | CXP | Screening RT (min) | Quantitative RT (min) |
|---|---|---|---|---|---|---|---|---|
| (V) | (eV) | (V) | ||||||
| Tetracycline | 445.5 | 410.3 | 100 | 30 | 30 | 3 | 13,3 | 9.77 |
| 445.5 | 427.5 | 30 | 5 | |||||
| Oxytetracycline | 461.3 | 201.1 | 100 | 30 | 30 | 6 | 12,9 | 9.66 |
| 461.3 | 443.1 | 50 | 3 | |||||
| Doxycycline | 445.4 | 428.3 | 100 | 40 | 30 | 4 | 18,5 | 10.4 |
| 445.4 | 201.3 | 50 | 3 | |||||
| Chlortetracycline | 479.2 | 444.2 | 100 | 30 | 40 | 5 | 13 | 10.26 |
| 479.2 | 462.2 | 10 | 5 | |||||
| Sulfaphénazole (IS) | 315.4 | 156 | 100 | 50 | 30 | 5 | 16.5 | |
| Demeclocycline (IS) | 465 | 448 | 100 | 45 | 18 | 3 | 9.99 |
Table 1: The MS/MS parameters of the selected antibacterial drugs.
DP: Declustering Potential; CE: Collision Energy; CXP: Collision cell exit potential; RT: Retention Time. Table 1: The MS/MS parameters of the selected antibacterial drugs.
The injection volume was 25µL for the two methods. Between the injections of standards and of samples and between injections of each sample, a blank (Water/Acetonitrile, V/V) injection was given.
Mass spectrometric analysis was carried out using electrospray ionization (ESI) in the positive mode. ESI parameters were capillary voltage (IS) of 5500 V, entrance potential (EP) of 10 V and source temperature of 700°C. The curtain gas was at 20psi and CAD gas was at level medium. Nebulizer gas (GS1) and auxiliary gas (GS2) were set at 40 psi and 50 psi, respectively. The specific MS/MS parameters for each target analyte are shown in Table 1.
**Validation Procedure**
Before the confirmation step, a qualitative method is implemented to monitor the presence or absence of tetracyclines residues in a sample and identification of the analytes.
According to European Commission Decision 2002/657/EC [11], the ratio of the chromatographic retention time of the analyte to that of the internal standard, i.e. the relative retention time of the analyte, should correspond to that of the calibration solution with a tolerance ± 2.5% for liquid chromatography (LC). Validated Excel® sheets were used to assess qualitative and quantitative criteria. Finally, stability of the standard solutions was based on the Fougères Laboratory studies. Thus, the stability of the stock solutions in methanol is 6 months below -18°C.
Qualitative Method
The validation was conducted according to European Commission Decision 2002/657/EC [11] and guidelines for the validation of screening methods [10]. Detection capability (CCβ) is assessed by using fortified samples according the MRL value (100µg/kg), with tetracyclines (Oxytetracycline, tetracycline, chlortetracycline, and doxycycline) for a screening purpose. Thus, CCβ is determined as the lowest concentration for which 20 measurements give less than 5% false-negative results [12].
In this study, the threshold value (T-value), the cut-off factor (Fm), detection observed limit (LOD), sensitivity and specificity were studied [10, 11].
For this, two series of samples are prepared, each one including seven samples. The first series will serve as “negative controls”, while the second series is supplemented with the analytes of interest (Quality control QC), at the target concentration (Cval = 0.5MRL). This pattern was repeated for three days. This generates 42 results allowing the calculation of the values T and Fm according to Equation 1 and Equation 2.
$$T = B + 1.64 \times SD_B$$
$$Fm = M - 1.64 \times SD$$
The means (B and M) and the standard deviations (SD$_B$ and SD) are calculated, from the responses obtained, at the level of the negative controls and the fortified samples respectively.
According to European Commission Decision 2002/657/EC [11], the suitable sensitivity of a screening method is demonstrated when the CCβ is below or equal to the MRL. Therefore, the false-compliant rate is below or equal to 5% at the MRL level. Consequently, CCβ value is validated only when Fm> T. Otherwise (Fm < T), additional studies, at a concentration greater than the target concentration, are necessary.
Specificity is the percentage of negative results, found among the expected negative results (controls). While the sensitivity is the percentage of positive results found among the expected positive results (QC).
A sensitivity greater than 95% was considered satisfactory. Detection limits (LOD) were calculated from the 21 QC samples according to Equation 3.
$$LOD = Cval \times H_{3 \times B} / M$$
LOD and Cval are in µg / kg, $H_{3 \times B}$ is the response (peak height) corresponding to 3 times the average noise and M is the average of the responses of the 21 supplemented samples. The noise is determined graphically and corresponds to the highest noise level, within the 1min MRM detection window.
Quantitative Method
The validation of a quantitative method was achieved according to European Commission Decision 2002/657/EC [11]. Therefore, the matrix matched standard calibration curve was constructed as described in sample preparation section. The efficiency of the analytical method was assessed by investigating the selectivity, matrix effect, linearity, trueness, repeatability and applicability. The quantitative criteria assessed were: regression model fit, trueness, repeatability, within-laboratory reproducibility and expended measurement uncertainty. The intermediate precision and the repeatability were expressed with a relative standard deviation (RSD) and trueness was evaluated using the recovery rate or bias.
The expended measurement uncertainty was assessed as a combined standard measurement uncertainty (Uc), which is determined as a root sum of squares of a standard deviation s characterising the precision of the measurement (repeatability, intermediate precision) and an estimate b accounting for measurement bias error [13]. Therefore, the equation to calculate Uc is given by Equation 4.
$$U_c = \sqrt{s^2 + b^2}$$
The confirmation of the analytes is based on the verification of the concentration calculated on the overall curve compared to the decision limit CCa; the relative intensity of the ionic ratio of the two transitions (MRM2 / MRM1); the relative retention time (referred to the IS tr) and the ratio of signal and background noise (S / N> 3).
Regarding the quantitative approach, the quantitation of the residue marker of tetracyclines required the analysis of both of the analyte of interest and its metabolite. In this case, three metabolites were added for this purpose: 4-epioxytetracycline, 4-epitetracycline and 4-epichlortetracycline. Then, seven analytes were optimized to perform a quantitative method.
**Samples Collection at the Slaughterhouse Level**
This is a cross-sectional descriptive study with an analytical aim. It is based on the evaluation of the residue levels of tetracyclines in chicken meat.
Sampling of chicken at slaughter points in the Algiers region was exhaustively carried out. The chicken were randomly chosen, with a minimum of five animals per slaughterhouse. Animals’ age was 60 days, according to the health certificate issued by veterinarians. In total, 151 chicken meat samples were collected at the slaughterhouse level in 2017. Samples were collected then analyzed by LC-MS/MS using our validated method.
The conformity of samples has been verified in a first time by comparing the responses obtained for each sample relative to the QC analyzed the same day. Secondly, the samples having exceeded the QC signal are analyzed with a confirmation and quantification method. Indeed, the ratio of the surfaces of the sample and of the QC was calculated for all the suspects. Only reports that exceeded unity (value of 1) were retained for confirmation and quantification.
**Results and Discussion**
**Optimization of Extraction and Clean-Up**
Concerning the optimization of the extraction of tetracyclines for a qualitative method, a simple and fast extraction procedure was performed according to the Fougères Laboratory method, where acetonitrile was used as a solvent for extraction. This is interesting when a multi-residues analysis method is to achieve. Indeed, that procedure was used for identification of twenty seven analytes by LC-MS/MS.
As widely described in literature, tetracyclines likely bind to metallic ions, consequently Na$_2$EDTA dihydrate were added specially to improve their extraction from the meat matrix. Thereby, an extraction was made using this salt before clean-up. In that way, the extraction was performed for the quantitative method.
**Optimization of the Analytical Method**
MS/MS data acquisition was performed in MRM (Multiple Reaction Monitoring) mode, where two product ions were chosen for each analyte. This was achieved by infusing 0.5µg/mL1 of individual standard solutions directly into the MS/MS system. The precursor ions of all analytes were [M + H]$^+$. Precursor and product ions, declustering potential (DP), collision energy (CE) and collision cell exit potential (CXP), retention times (RT) values are shown in Table 1. The best sensitivity for all drugs was determined to provide the highest signals for identification, quantification and confirmation.
The MS/MS optimization results show the loss of water or ammonia after fragmentation the analytes studied. This is due to their structure, which is formed by an octahydrotetracene-2-carboxamide skeleton [14].
The selectivity was assessed by examination the chromatograms. We observed that no interference was possible with the three analytes, their metabolites and doxycycline, since the diagnostic ions obtained do not have the same masses. However, doxycycline precursor ion and tetracycline precursor ion were identical, but the difference in polarity between the two analytes and the reversed abundance of the product ions made it easier for us to distinguish the two analytes. In fact, doxycycline being the least polar is eluted last among the four tetracyclines after chlortetracycline. The latter is preceded by tetracycline. Oxytetracycline is the first to be detected.
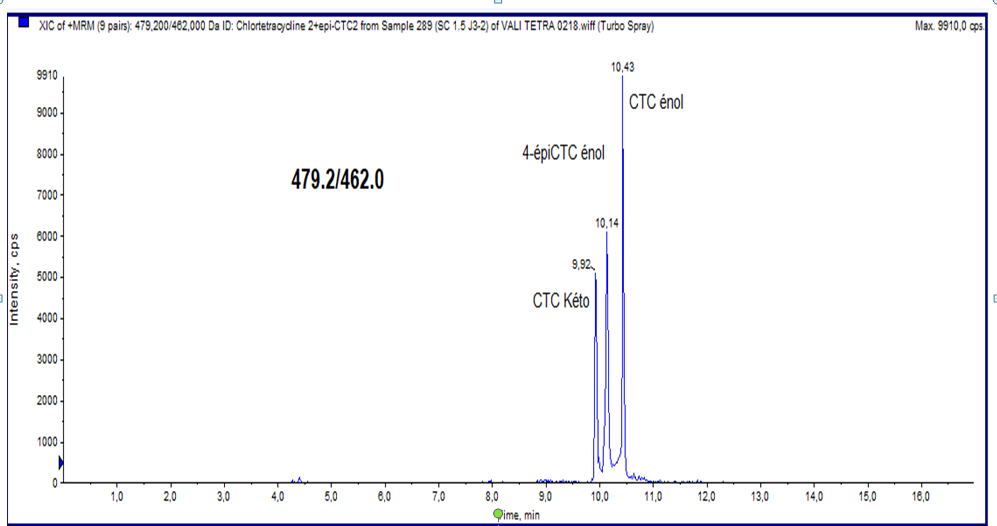
Else ways, the analysis of chlortetracycline, by LC-MS/MS under these conditions, confirms a phenomenon widely described in the literature, which is the tautomerization of chlortetracycline and its epimer. These studies investigated the keto-enol forms of the CTC and its epimer 4-epi-CTC in tissues (Figure 2). The 6-iso-CTC metabolite was also first described in these studies, but only in eggs and not in tissues. All of these forms of CTC are isobaric compounds and cannot be distinguished by their specific mass [15].
Ion pairing reagents can influence the signal of the target compound in the electrosprayed liquid [16]. For this purpose, heptafluorobutyric acid (HFBA) and pentafluoropropionic acid (PFPA) were previously used as ion pairing reagents diluted in mobile phase. However, a lack of repeatability of retention times (more than a minute) was observed. Thus, we have opted for the use of trifluoroacetic acid, which allowed us to obtain satisfactory results. Chromatographic and spectrometric conditions were described in Materials and Methods section.
The qualitative method had a total run time of 25 min and all analytes eluted within 18.5 min. The shortest retention time was observed with oxytetracycline (12.84 min), which is explained by a highest affinity with the aqueous phase and a lowest interaction with the stationary phase. On the other hand, the longest retention time was observed with doxycycline (18.55 min).
Extracted ion chromatograms from liquid chromatography−tandem mass spectrometry on chicken meat samples, obtained from the qualitative method.
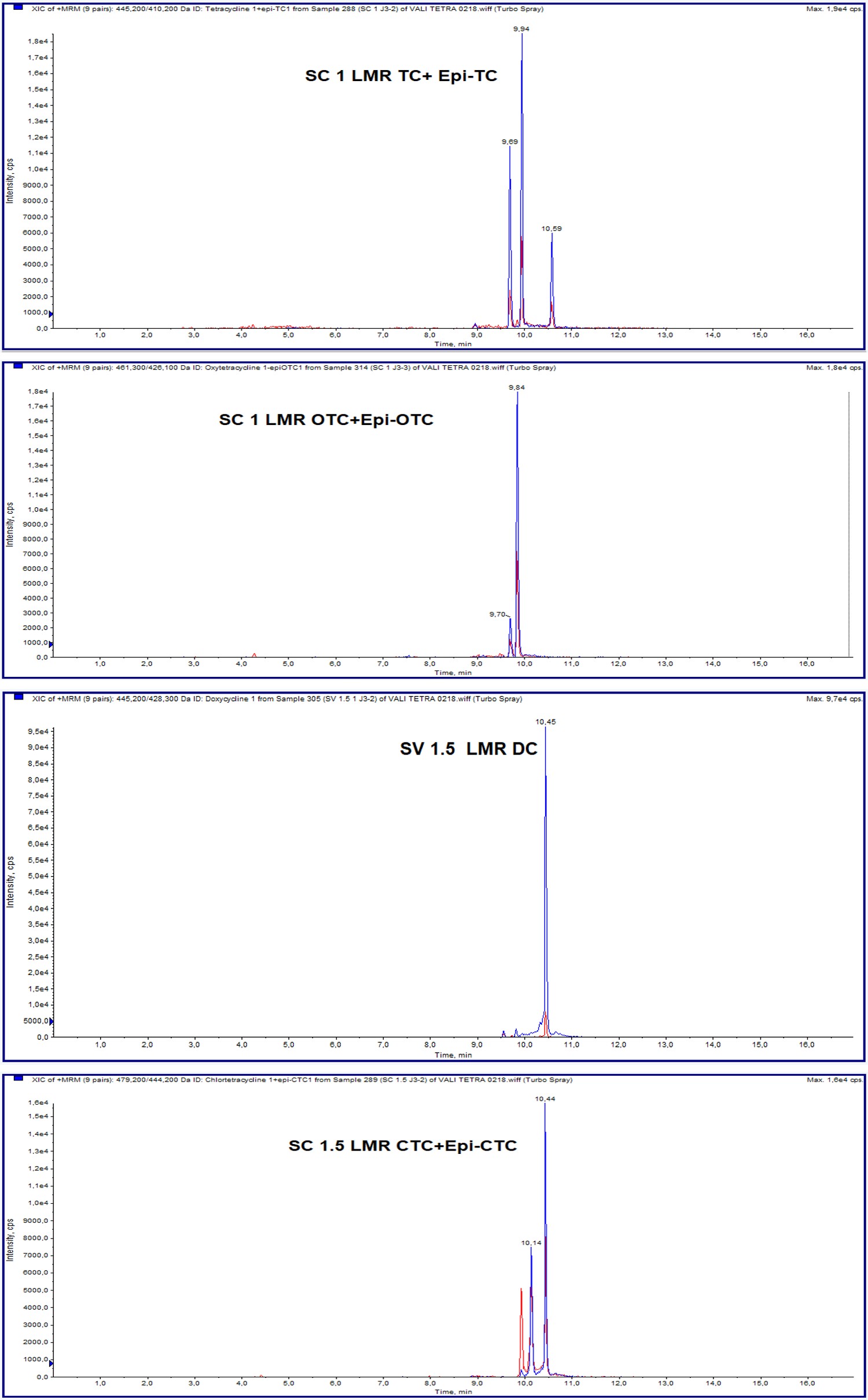
The total time of run was 18 min for the quantitative method. All analytes eluted within 11min. The first analyte eluted was oxytetracycline at 9.86min and the last one was doxycycline at 10.60min. On the other hand, analytes were eluted after their epimers. Tetracyclines and their metabolites chromatograms acquired from the quantitative method.
Validation
The goal of this work was to identify and quantify tetracyclines residues in raw chicken meat using the LC- MS/MS method. Each analyte was identified unequivocally by the presence of tow transitions eluted at the same time. Indeed, in quantitative method, the first chromatographic peak is used for the quantification and the second for the confirmation. On the other hand, these two peaks allow the identification and the confirmation as regards the qualitative method. The identification of sulfonamides and quinolones antimicrobials residues in meat explains the long run time in this method.
Qualitative Method Validation Results
As described in Materials and Methods section, qualitative validation criteria were evaluated according to EC Decision 2002/657/EC [11]. Therefore, sensitivity, CCß, Fm and T values were assessed as shown in Table 2. The Cval corresponds to half of the MRL value of each analyte (50µg/ kg).
| Analytes | MRM 1 | MRM 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fm | T | CCß | Se | LODe | Fm | T | CCß | Se | LODe | |
| Tetracycline | 8852,84 | 1722,31 | ˂Cval | 100% | 21,87 | 821,42 | 214,31 | ˂Cval | 100% | 32,57 |
| Oxytetracycline | 9258,50 | 4790,06 | ˂Cval | 95% | 31,55 | 2719,86 | 1398,50 | ˂Cval | 100% | 32,73 |
| Doxycycline | 9218,94 | 5841,37 | ˂Cval | 95% | 13,25 | 4384,15 | 400,58 | ˂Cval | 100% | 42,58 |
| Chlortetracycline | 10243,23 | 7600,76 | ˂Cval | 95% | 16,84 | 6817,74 | 1373,85 | ˂Cval | 100% | 82,06 |
Table 2: CCß, Fm, T-value and sensitivity of tetracyclines in chicken meat.
MRL =100 µg/kg, Cval = 50 µg/kg, Se: Sensitivity, LODe: estimated LOD (µg/kg). Table 2: CCß, Fm, T-value and sensitivity of tetracyclines in chicken meat.
The specificity of the method was verified by analyzing twenty samples of chicken breasts from different origins and the specificity was 100% for all analytes because no peak was detected in these samples at the retention time corresponding to each analyte.
The criterion of the relative retention time was met for all validation standards. The variability of the relative retention time of the four analytes was satisfactory with RSD values between 0.36 and 1.86%.
The limit of detection estimated were shown in Table 2. The determined LOD was below than 50 µg/kg for both transitions for all analytes, except for chlortetracycline for MRM2 for which LOD was out of Cval value.
Quantitative Method Validation Results
Several criteria were checked for each of the selected transitions: specificity, retention time, relative ion intensity tolerances and signal-to-noise ratio, in terms of identification.
Specificity was checked not only by examining the chromatograms of different matrix control samples at the target retention times but also by injecting different target analytes to verify that there was no interference between monitored MRM transitions.
The chromatograms of different matrix control samples was examined at the target retention times and results were satisfactory. Indeed, tetracyclines analysis was specific for oxytetracycline, chlortetracycline and tetracycline. However, the specificity was to a lesser extent for Doxycycline.
Therefore, the programming of the four samples of water/ ACN solutions between injections proved to be essential to address this contamination.
The ratio between the two identification transitions (relative ion intensities) were found to be within the maximum permitted tolerances for all validation standards except two, for doxycycline and 4-epioxytetracycline (Table 3).
| Compound | VS | Recovery (%) | Precision (%) | Interday precision (RSD %) | Relative ion intensity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Concen tration | |||||||||||
| (µg/kg) | Trueness | Repetability | Within-lab. | Uncertainty | Day 1 | Day 2 | Day 3 | Tole rance | |||
| (%) | (%) | reproduc ibility (%) | (%) | ||||||||
| Oxytetracycline | 50 µg/kg | 92,3 | 16,5 | 20 | 39,75 | 27,28 | 51,20 | 9,4% | 5,5% | 4,7% | 25% |
| 100 µg/ kg | 94,3 | 12,5 | 14 | ||||||||
| 150 µg/kg | 86,8 | 13 | 25,6 | ||||||||
| 4-epioxytetra cycline | 50 µg/kg | 90,8 | 19 | 25 | 49,26 | 62,83 | 70,64 | 28,5% | 24,8% | 18,6% | 25% |
| 100 µg/ kg | 104,6 | 15 | 31 | ||||||||
| 150 µg/kg | 88,3 | 21 | 35 | ||||||||
| Doxycycline | 50 µg/kg | 103,5 | 39 | 48 | 95,60 | 152,3 | 135,34 | 5,4% | 71,3% | 5,8% | 50% |
| 100 µg/ kg | 134,7 | 43 | 76 | ||||||||
| 150 µg/kg | 102,6 | 22 | 68 | ||||||||
| Tetracycline | 50 µg/kg | 102,6 | 21 | 22 | 44,31 | 39,30 | 60,12 | 12,2% | 14,5% | 12,5% | 25% |
| 100 µg/ kg | 97,5 | 14 | 20 | ||||||||
| 150 µg/kg | 76,6 | 13,5 | 30 | ||||||||
| 4-epitetracycline | 50 µg/kg | 103,0 | 26 | 26 | 52,00 | 47,74 | 67,60 | 12,2% | 10,4% | 8,5% | 25% |
| 100 µg/ kg | 104,8 | 15 | 24 | ||||||||
| 150 µg/kg | 85,5 | 17 | 34 | ||||||||
| Chlortetracycline | 50 µg/kg | 87,5 | 20 | 23,5 | 46,9 | 38 | 62,6 | 7,3% | 6,3% | 7,7% | 20% |
| 100 µg/ kg | 84,7 | 16 | 19,0 | ||||||||
| 150 µg/kg | 81,3 | 15,0 | 31 | ||||||||
| 4-epichlortetracycline | 50 µg/kg | 110,7 | 13,5 | 21 | 42,16 | 28 | 32,78 | 7,9% | 13,3% | 11,0% | 20% |
| 100 µg/ kg | 99,6 | 9,5 | 14 | ||||||||
| 150 µg/kg | 90,0 | 8 | 16 |
Table 3: Trueness, Repeatability and within-Laboratory Reproducibility results.
The signal-to-noise ratio was always above 3 for both transitions on each day and for all compounds.
A simple linear regression model was chosen for all analytes. Calibration curves were satisfactory with determination coefficient (r2) between de 0.91 à 0.98%, where the lowest values were observed for doxycycline.
The quantitative performance of the method results in terms of trueness, precision and expended uncertainty are enumerated in Table 3. The quantitative results were compliant with the requirements of Decision 2002/657/ EC except for doxycycline for which, even though trueness results were acceptable, repeatability and reproducibility RSD was too high. In the aim to achieve better results, the use of an isotopically labeled standard apparently was indicated, not only of doxycycline Gaugain M, et al. [17], but also for the three tetracyclines. This may reduce the matrix effect, which is not accurately corrected by the internal standard demeclocycline [17].
As described in Decision 2002/657/EC, the decision limits (CCα) were assessed and were between 107 and 141 on three days, except for doxycycline, 4-epioxytetracycline and chlortetracycline, for which CCα values (for one day), were higher than the maximal CCα expected (144µg/kg).
Globally, the validation results were in agreement with an exposure assessment purpose, even though some values were out of the maximum permitted tolerances in terms of quantitative validation criteria.
Assessment of Tetracyclines Residues in Chicken Meat Samples
Qualitative Assessment
The analysis of 151 samples revealed the presence of oxytetracycline or doxycycline alone or both of them in twenty-five (16.55%) samples. Then, oxytetracycline was identified in thirteen (8.60%) samples, probably due to its greater use in cattle than in poultry. Apart from that, doxycycline was detected in fifteen (9.93%) samples. The withdrawal time for doxycycline in chickens is five days. This predicts its presence in chicken samples in addition to its more pronounced lipophilic character compared to other tetracyclines. Three samples contained both oxytetracycline and doxycycline.
Quantitative Assessment
Five among the 25 positive samples were contaminated with oxytetracycline or doxycycline at a level higher than the MRL value (100µg/kg). The surfaces sample/QC ratio was between 1.29 and 3.48. Therefore, calibration and validation standards were analyzed simultaneously with the unknown chicken meat. The calibration results in terms of relative intensity, relative retention time, CCα, signal to noise, are provided in Table 4.
| Concentration Level | Relative intensity (MRM /MRM ) 2 1 | Relative retention time (min) | S/N (MRM ) 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (µg/kg) | DC | OXT | Epi-OXT | DC | OXT | Epi-OXT | DC | OXT | Epi-OXT |
| 50 | 0,09 | 0,42 | 0,65 | 1,06 | 0,96 | 0,95 | 205 | 330 | 88,3 |
| 0,09 | 0,33 | 0,89 | 1,04 | 0,98 | 0,98 | 352 | 171 | 92,6 | |
| 100 | 0,08 | 0,34 | 0,40 | 1,06 | 0,97 | 0,95 | |||
| 0,08 | 0,34 | 0,51 | 1,04 | 0,98 | 0,98 | ||||
| 150 | 0,08 | 0,35 | 0,43 | 1,06 | 0,97 | 0,95 | |||
| 0,09 | 0,38 | 0,56 | 1,04 | 0,98 | 0,98 | ||||
| 200 | 0,09 | 0,32 | 0,43 | 1,07 | 0,97 | 0,96 | |||
| 0,08 | 0,34 | 0,58 | 1,04 | 0,98 | 0,97 | ||||
| Mean | 8,5% | 35,4% | 55,7% | 1,05 | 0,97 | 0,97 | |||
| Tolerance | 50% | 25% | 20% | 2,5% | |||||
| Max | 12,7% | 44,3% | 66,8% | 1,08 | 1,00 | 0,99 | |||
| Min | 4,23% | 26,6% | 44,5% | 1,03 | 0,95 | 0,94 | |||
| CC α (µg/kg) | 140 | 123 | 134,5 |
Table 4: Relative intensity, relative retention time, CCα and signal to noise of sample analysis.
Both transitions exist in all samples with a signal-to-noise ratio significantly greater than 3. Three samples contained 86.49, 97.23 and 120.16 µg/kg of sum of oxyetracycline and its metabolite, 4-epioxytetracycline. Even though this residue contamination is significant considering the MRL value (100µg/kg), these values do not exceed the corresponding CCα values (130.15, 129.51 and 130.46 µg/kg respectively). In addition, the same finding was observed with doxycycline, which was quantified at 46.8, 91.65 and 124.24 µg/kg with decision limits value of 140 g/kg. Consequently, the five samples were compliant according to the decision limit values.
Conclusion
Qualitative and quantitative methods were successfully validated for the detection and quantification tetracyclines by LC-MS/MS. Thus, a simple, cheap, fast, reliable and sensitive multiresidues analytical method was developed for the simultaneous determination of tetracyclines in chicken muscle samples using simple liquid extraction with LC-MS/ MS.
In addition, quantification criteria as required in Decision 2002/657/EC were met for oxytetracycline, tetracycline, chlortetracycline and their metabolites and doxycycline.
The developed method was effectively applied to real samples, and the results of tetracyclines residues showed that oxytetracycline and doxycycline were identified in twenty-five chicken meat samples. The comparison with the QC sample allowed us to confirm that five samples were contaminated with an amount over the MRL value. Therefore, quantification of oxytetracyclin, its metabolite and doxycycline in five samples revealed that they were compliant according the Decision 2002/657/EC. However, additional studies with a larger sample size are necessary to confirm tetracyclines residues in chicken muscles to assure food safety.
References
-
Mungroo NA, Neethirajan S (2014) Biosensors for the Detection of Antibiotics in Poultry Industry—A Review. Biosensors 4(4): 472-493.
-
Klein EY, Van Boeckel TP, Martinez EM, Pant S, Gandra S, et al. (2018) Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proceedings of the National Academy of Sciences of the United States of America 115(15): E3463–E3470.
-
Méheust D, Chevance A, Moulin G (2016) Suivi des ventes de médicaments vétérinaires contenant des antibiotiques en France en 2015. Rapport annuel ANSES pp: 1‑106.
-
Matzov D, Bashan A, Yonath A (2017) A Bright Future for Antibiotics? Annual review of biochemistry 86: 567-583.
-
Marshall BM, Levy SB (2011) Food animals and antimicrobials: impacts on human health. Clinical microbiology reviews 24(4): 718-733.
-
Goucem R (2015) L’antibiothérapie en aviculture et ses implications. ENSV d’Alger.
-
Agroligne (2017) Usages des antibiotiques en élevage Quels risques pour la santé animale et humaine ? L’essentiel de l’agroalimentaire et l’agriculture - N°104 9: 60.
-
European Commission Decision (2009) Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substance and their classification, regarding maximum residue limits in foodstuffs of animal origin. Official Journal of the European union.
-
Algerian Ministry of Commerce (2016) Arrête interministériel du 20 juin 2016 fixant les listes ainsi que les limites maximales de résidus de médicaments vétérinaires ou de substances pharmacologiquement actives tolérées dans les denrées alimentaires d’origine animale.
-
European Commission (2010) Guidelines for the validation of screening methods for residues of veterinary medicines (initial validation and transfer).
-
European Commission (2002) Decision 2002/657/ EC of implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Ecolex.
-
Pikkemaat MG (2009) Microbial screening methods for detection of antibiotic residues in slaughter animals. Analytical and bioanalytical chemistry 395(4): 893-905.
-
Gaugain M, Raynaud A, Bourcier S, Verdon E, Hurtaud- Pessel D (2021) Development of a liquid chromatography- tandem mass spectrometry method to determine colistin, bacitracin and virginiamycin M1 at cross-contamination levels in animal feed. Food additives contaminants Part A Chemistry analysis control exposure risk assessment 38(9): 1481-1494.
-
Susakate S, Poapolathep S, Chokejaroenrat C, Tanhan P, Hajslova J, et al. (2019) Multiclass analysis of antimicrobial drugs in shrimp muscle by ultra-high performance liquid chromatography-tandem mass spectrometry. Journal of food and drug analysis 27(1): 118-134.
-
Gaugain M, Gautier S, Bourcier S, Jacques AM, Laurentie M, et al. (2015) 6-Iso-chlortetracycline or keto form of chlortetracycline? Need for clarification for relevant monitoring of chlortetracycline residues in food. Food additives & contaminants Part A 32(7): 1105-1115.
-
Holcapek M, Volná K, Jandera P, Kolárová L, Lemr K, et al. (2004) Effects of ion-pairing reagents on the electrospray signal suppression of sulphonated dyes and intermediates. Journal of mass spectrom 39(1): 43-50
-
Gaugain M, Fourmond MP, Fuselier R, Verdon E, Roudaut B, et al. (2020) Control of Antimicrobials in Feed Using Liquid Chromatography−Tandem Mass Spectrometry: Assessment of Cross-Contamination Rates at the Farm Level. Journal of Agricultural and Food Chemistry 68(34): 9033-9042.
- Evaluation of Skin Aging Preventive Effects of Cherry Blossom Petal Extracts Through Antioxidant and Anti-Glycation Activities
- Is Cell Death Responsible for False Positive Results of In Vivo Comet Assay?
- Pattern of Gonadal Hormones in Oral Testosterone-Supplimented Male Wistar Rats with Diabetes-Induced Hypogonadism
- Re-Evaluation of the Genotoxicity of Currently Used Food Dyes in Mouse Multiple Organs Via Continuous Administration by Drinking Using the Comet Assay
- Pharmacogenetics of Type 2 Diabetes Mellitus: Linking Genetic Variability to Drug Efficacy and its Cardiovascular Outcomes
- Exploratory Proteomic Profiling of SARS-CoV-2 Infected THP-1 Macrophages Reveals Alterations in Inflammatory Response and Cellular Metabolism