Combination Therapy of Bispecific Protein of Anti-PD-L1 Fused with TGF- β Receptor in Cancer
Transforming growth factor β (TGF-β) and programmed death-ligand 1 (PD-L1) initiate signaling pathways with complementary, nonredundant immunosuppressive functions in the tumor microenvironment (TME). In the TME, dysregulated TGF-β signaling suppresses antitumor immunity and promotes cancer fibrosis, epithelial-mesenchymal transition, and angiogenesis. Meanwhile, PD-L1 expression inactivates cytotoxic T cell function and restricts immunosurveillance in the TME. Anti-PD-L1 therapies have been approved for the treatment of various cancers, while TGF-β signaling in the TME is associated with resistance to anti-PD-L1 therapies. Here, we have reviewed the rationale of the TGF-β and PD-L1 pathways in cancer and discussed current strategies using combination therapies that block these pathways separately or approaches with dual-targeting agents that may block the pathways simultaneously. Importantly, according to current clinical results, we propose the developing strategy of combination treatment with two or more agents.
Dual Targeting of Transforming Growth Factor β (TGF-β) and Programmed Death Ligand 1 (PD-L1) in Cancer.
TGF-β Signaling in Cancer
TGF-β is a pleiotropic cytokine comprising three isoforms: TGF-β1, TGF-β2, and TGF-β3. They share nearly 70% homology and perform similar biologic activities. The activity of TGF-β is regulated at a variety of steps [1, 2]. TGF-β is secreted in an inactive form attached to a peptide partner the latency-associated peptide (LAP). This latent form can undergo isoform-specific activation through the cleavage of the LAP by extracellular proteases to release active TGF-β. The release of active TGF-β could induced by a physical force via integrins, such as αvβ6 or αvβ8, binding to the LAP or by the modification of the LAP through thrombospondin [3]. The active TGF-β then binds to the serine/threonine protein kinase receptors TGF-βRI and TGF-βRII, sometimes with the aid of the accessory co-receptor β-glycan; the activated receptor complex then phosphorylates specific signal- transduction protein SMADs such as SMAD2, SMAD3 and SMAD4. The phosphorylated SMADs then form heterotrimeric SMAD complexes, translocate into the nucleus, interact with cell-specific transcription factors, and induce downstream gene expression [3]. While SMAD2, SMAD3, and SMAD4 are required in TGF-β signaling, SMAD6 and SMAD7 inhibit TGF-β signaling [3]. The resulting signaling pathway can induce a large and diverse set of cellular responses that are highly context and tissue specific [3, 4].
Physiologically, TGF-β maintains immunological self-tolerance and acts to suppress cancer by regulating epithelial proliferation, apoptosis, and differentiation [3, 5]. TGF-β signaling undergoes changes during malignant transformation, resulting in TGF-β functioning as a tumor promoter rather than a suppressor [4, 6]. Aberrant TGF-β activation and signaling can promote disease progression by stimulating epithelial to mesenchymal transition (EMT) and angiogenesis, activating cancer-associated fibroblast (CAF), and enhancing immunosuppression within the tumor microenvironment (TME) [7, 8, 9]. High expression of TGF-β in the TME correlates with poor clinical outcome and increased likelihood of metastasis in various tumor types [10, 11].
EMT is involved in migration and invasion of cancer cells [12]. Increased cancer stem cells are observed in EMT [13], while TGF-β signaling further promote cancer stem cells via enabling epithelial cells acquiring stem cell-like properties [14]. EMT is also associated with resistance to anti-programmed cell death 1 (PD-1) and anti- programmed cell death ligand 1 (PD-L1) therapies, targeted therapies, and chemotherapy [15, 16, 17]. Aberrant TGF-β signaling can upregulate the expression of proangiogenic factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF), resulting in increased blood vessel density and tumor size [18, 19]. TGF-β signaling in the TME is associated with the transition of fibroblasts into CAFs that contribute to drug resistance and metastasis of tumors [20, 21]. CAFs remodel the extracellular matrix to influence T-cell migration and trap T cells in the stroma. CAFs express a variety of cytokines, such as interleukin (IL)-6, IL-8, IL-11, CCL2, PGE2, CXCL12, and TGF-β, which are involved in immune suppression and metastasis [17, 20, 21, 22, 23].
TGF-β plays an important role in immunity, particularly in immunosuppression, since most immune cells respond strongly to its effects [4]. Immunosuppression by TGF-β in cancer involves a phenotypic change in a variety of immune cells such as dendritic cells, tumor-associated macrophages (TAMs), tumor-associated neutrophils, natural killer (NK) cells, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Treg cells), and cytotoxic T cells [8, 9, 23, 24]. TGF-β is an autocrine survival signal for myeloid precursors and drives their differentiation to highly immunosuppressive MDSCs at the expense of macrophages and dendritic cells [25]. In the TGF-β-rich TME, dendritic cells shift into a tolerogenic phenotype, with reduced antigen presentation and ability of activating T cells [26]. Macrophages shift from an inflammatory (M1) to a tumor-trophic (M2) phenotype, becoming Tumor-associated macrophages. This phenotype reduces the expression of pro-inflammatory cytokines, while expression of TGF-β and VEGF are increased [27]. Mature neutrophils in the TME preferentially adopt a phenotype that promotes tumor growth, immunosuppression, metastasis, invasion, and angiogenesis [28]. TGF-β can suppress the proliferation and cytotoxicity activity as well as the production of interferon gamma (IFNγ) of NK cells. As we know, IFNγ is an critical cytokine for the activation of macrophages, NK cells and neutrophils [3, 24]. Undifferentiated T cells can switch to a Treg phenotype in the presence of TGF-β; this switch may lead to both the inactivation of effector and cytotoxic T cells and an increase function of MDSCs [3, 24]. Systemic TGF-β levels are often increased in people with cancer relative to those in healthy individuals, and elevated TGF-β levels are associated with poor prognosis and aggressive cancer [27]. However, the use of TGF-β as a biomarker is complicated by the technological challenges associated with measuring active TGF-β levels in the TME and the predominance of latent TGF-β in circulation [29].
TGF-β in Cancer Therapy
Inhibition of the TGF-β pathway remains an active area of interest in cancer research. Antibodies, vaccines, antisense oligonucleotides, and small-molecule inhibitors against TGF-β pathway have all been investigated in clinical trials of solid tumors [1, 8]. In addition to overcoming immunosuppression, preclinical studies have demonstrated that the blockade of TGF-β signaling suppresses fibrosis, EMT, angiogenesis, and tumor growth [7, 8].
There are three major ways for targeting TGF-β: blocking the expression and activation of TGF-β, blocking the binding of TGF-β to its receptors (including sequestering or “trapping” TGF-β), or inhibiting TGF-β receptor kinase signaling. Therapies that block TGF-β activation and expression include gemogenovatucel-T, a plasmid-based therapy; SRK-181, a TGF-β1 antibody; abituzumab, a pan-av integrin antibody; PF-06940434, an avβ8 integrin antagonist; and cotsiranib, a small interfering RNA therapeutic that inhibits the expression of TGF-β1. Gemogenovatucel-T is a gene therapy consisting of a plasmid of a bifunctional short hairpin RNA that suppresses mature TGF-β1/2 processing and expresses the immune- stimulatory cytokine granulocyte-macrophage colony- stimulating factor (GM-CSF) (NCT03073525) [30]. In a phase 1 prospective study, patients receiving gemogenovatucel-T (n=15) had one year overall survival (OS) rate of 73% vs 23% in patients not receiving gemogenovatucel-T (n=13) [31]. No grade 3 treatment-related adverse events (TRAEs) were observed in the gemogenovatucel-T-treated patients, and the most common TRAEs included grade 1 injection site induration and injection site erythema reported in 12 and 11 patients, respectively. SRK-181, which inhibits TGF-β1 activation by targeting the regions of latent TGF-β complex that mediate cell-associated activation, may provide a means to selectively block activity in certain cells and it is currently being studied in patients with solid tumors (NCT04291079)
[32]. Abituzumab blocks the activation of TGF-β by binding to the integrin αv subunit. In a phase 1/2 trial in patients with metastatic colorectal cancer (N=216), abituzumab therapy in combination with the standard of care (SOC) showed acceptable tolerability, but the primary endpoint of progression-free survival was not met. However, OS benefit was observed in patients with tumors showing high integrin αvβ6 expression in each of the abituzumab dosing arms compared with the SOC; the median OS was not reached (NR) (95% CI, 9.7-NR months) for the abituzumab 1000 mg + SOC group, 15.0 months (95% CI, 10.5-23.2 months) for the abituzumab 500 mg + SOC group, and 10.2 months (95% CI, 5.8-13.1 months) for the SOC group [33]. Though high integrin αvβ6 expression was correlated with worse survival outcomes in the SOC treatment group, it was correlated with increased survival benefit upon treatment with either abituzumab 1000 mg + SOC (hazard ratio [HR], 0.41 vs 1.58 in the high integrin and low integrin groups, respectively) or abituzumab 500 mg + SOC (HR, 0.55 vs 1.48 in the high integrin and low integrin groups, respectively). Treatment- emergent AEs (TEAEs) occurred in 100% of patients in both the lower-dose and the higher-dose abituzumab groups, with the most common TEAEs being diarrhea (65% and 67 %, respectively), stomatitis (25% and 30%), and asthenia (21% and 29%). PF-06940434 is currently under investigation for treatment of patients with advanced or metastatic solid tumors, but data indicating efficacy have yet to be published (NCT04152018). Cotsiranib, a TGF-β1 and COX-2 small interfering RNA inhibitor, is under investigation in basal cell carcinoma (NCT04669808) and in patients with advanced solid tumors with cholangiocarcinoma, hepatocellular carcinoma, or liver metastases (NCT04676633).
Therapies that binds to and inhibits the protein TGF-β currently in clinical development include neutralizing antibodies (fresolimumab, SAR439459, NCT03192345; and NIS793, NCT04390763) and a fusion protein that functions as a TGF-β1/3“rap” (AVID200, NCT03834662) [34]. The pan- TGF-β neutralizing antibody, fresolimumab, was investigated in patients with advanced melanoma or renal cell carcinoma (N=29); 1 patient experienced a partial response (PR), and 6 patients experienced stable disease. Gingival bleeding, headache, and epistaxis were the most common TRAEs, with each occurring in 13.8% of patients. TGF-β -related skin lesions, including actinic keratosis and hyperkeratosis (10.3% each), keratoacanthoma and squamous cell carcinoma of the skin (6.9% each), and basal cell carcinoma (3.4%), were also common [34]. SAR439459 is a pan-TGF-β antibody that has demonstrated preclinical activity in human cell lines and murine tumor models, in which treatment inhibited the TGF-β-induced suppression of CD8+ T cells and the development of Treg cells [35]. In a phase 1b study (NCT02947165), no dose-limiting toxicities were reported in patients (N=120) with advanced solid tumors receiving the TGF-β1/2 inhibitor, NIS793 in combination with anti-PD1 antibody spartalizumab, and PRs were reported in 4 patients across cohorts. The most common TRAEs were rash (n=15), pruritis (n=10), fatigue (n=9), and nausea (n=8) [36]. TGF-β inhibitor AVID200 in syngeneic mouse tumor models has demonstrated activity and increased T-cell infiltration into the tumor microenvironment [37], as well as antifibrotic activity in a separate model of idiopathic pulmonary fibrosis (Forbius official announcement). In idiopathic pulmonary fibrosis, the accumulation of activated, heterogenous myofibroblasts, analogous to the conversion of fibroblasts to CAFs in cancer, is mediated by TGF-β signaling [38]. In the phase 1 AVID200- 03 dose-escalation study (NCT03834662), proinflammatory serum marker levels were increased in a dose-dependent manner in patients (N=19) receiving AVID200; tumor biopsies showed modulation of TGF-β signaling and immune activation [39]. Grade 3 TRAEs were reported in 2 patients (diarrhea, lipase elevation, and anemia).
Another class is the oral small-molecule inhibitors of TGF- βR kinases, including LY3200882, vactosertib, LY2109761, and galunisertib, that prevent SMAD-mediated TGF-β signaling (NCT02937272) [40, 41, 42]. The TGF-βRI inhibitor, LY3200882, is currently being investigated in patients with solid tumors (NCT02937272). Vactosertib, a small- molecule kinase inhibitor of TGF-βRI, is being investigated in combination with anti-PD1 antibody pembrolizumab in colorectal cancer or gastric/gastroesophageal junction cancer (NCT03724851); vactosertib is also being investigated in combination with anti-PD-L1 antibody durvalumab in the second-line treatment of non-small cell lung cancer (NSCLC; NCT03732274). In a phase 1 dose-escalation study, 35.3% of patients receiving vactosertib at 2140 mg (n=17) achieved stable disease. The most common TRAE was fatigue [42]. In a preclinical study, the TGF-βRI inhibitor, LY2109761, depleted high CD44 and ld1 glioma-initiating cells (both indicators of poor prognosis) in human glioblastoma specimens [40]. In a translational study, the TGF-βRI inhibitor galunisertib was used as a monotherapy to enhance antitumor T-cell immunity and antigen spreading in a mouse model of breast cancer [41]. Clinically relevant doses of galunisertib were used to enhance the antitumor activity of anti-PD-L1 therapy (antimurine PD-L1 clone), resulting in tumor regression and enhanced T-cell activation in a murine colorectal cancer model [41].
TGF-β signaling has been implicated in the development and function of the heart, which may present a challenge for systemic inhibition of TGF-β as an anticancer therapy [43]. Toxicity concerns have been raised for inhibitors of the TGF-βRI kinase ALK5, based on preclinical studies that showed increased incidence of heart valve lesions in animals receiving the TGF-βRI kinase inhibitors AZ12601011 and AZ12799734 [44]. Galunisertib, which belongs to this class, was selected for clinical development because incidence of heart lesions appeared only at very high doses or with continuous treatment for 6 months. Clinical research involving >300 patients has shown that the animal model toxicities of concern for galunisertib have not been reported in humans when intermittent dosing is applied [45]. An anti-TGF-βRII antibody, LY3022859, was under clinical investigation in patients with advanced solid tumors (N=14), but the trial was discontinued due to the absence of clinical efficacy and incidence of cytokine storm, despite prophylactic administration of antihistamines and corticosteroids [46].
PD-L1 Signaling in Cancer
The PD-1 receptor and its ligand, PD-L1, are critically involved in tumor immunity. The PD-1/PD-L1 pathway mediates tumor immune evasion by suppressing cell killing of cytotoxic T cells and NK cells that express PD-1 via the expression of PD-L1 in tumor cells, Treg cells, MDSCs, and macrophages in the TME, resulting in loss of tumor immunosurveillance [47, 48]. Inhibition of the PD-L1 pathway, by blocking of either the receptor or the ligand, has great potential to disinhibit cytotoxic T cells in the TME, resulting in long-lasting antitumor activity in subsets of patients across different tumor types [47, 48, 49, 50]. However, releasing T cells from inhibition via anti-PD-1/PD-L1 therapy may initiate a negative feedback loop, stimulating PD-L1 production by MDSC and subsequent T-cell reinhibition [48, 50].
Anti-PD-1/PD-L1 therapy has changed the treatment landscape for a variety of solid tumor types, including NSCLC, melanoma, squamous cell carcinoma of the head and neck (SCCHN), renal cell carcinoma (RCC), and urothelial carcinoma (UC), due to higher response rates and more manageable toxicity profiles than chemotherapy. Prior to the use of anti-PD-1/PD-L1 therapy, second-line chemotherapy provided response rates of <10% in NSCLC [51]. In the phase 1 KEYNOTE-001 study of the PD-1 inhibitor pembrolizumab in patients with advanced NSCLC (N=495), the objective response rate (ORR) was 19.4%, with a median duration of response of 12.5 months. TRAEs were reported in 70.9% of patients (most commonly fatigue [19.4%], pruritis [10.7%], and decreased appetite [10.5%]). This led to the National Comprehensive Cancer Network recommending pembrolizumab as a second-line treatment for PD-L1- positive NSCLC (NCCN, Non-Small Cell Lung Cancer, Version 2.2021) [52]. Chemotherapy (dacarbazine) was the standard of care for metastatic melanoma for 3 decades after its US Food and Drug Administration approval. In the phase 3 CheckMate 066 study in patients with advanced melanoma (N=418), the PD-1 inhibitor nivolumab had a median OS of 37.5 months in the nivolumab group Vs 11.2 months in the dacarbazine group and an ORR of 40.0% (95% CI, 33.3%- 47.0%) vs 13.9% (95% CI, 9.5%-19.4%) with dacarbazine (odds ratio, 4.06), with 66.7% of nivolumab responders experiencing an ongoing response after 38.4 months [53, 54]. TRAEs were reported in 77.7% and 77.6% in the nivolumab and dacarbazine cohorts, respectively; the most common TRAEs were pruritis (23.8%), diarrhea (18.9%), and rash (18.4%) in the nivolumab cohort. Second-line chemotherapy provided response rates of 3% to 13% in patients with recurrent/metastatic SCCHN, but promising results from immune checkpoint inhibitors have led to the approval of pembrolizumab and nivolumab in this setting [55]. In the phase 1b KEYNOTE-012 study cohort of pembrolizumab in patients with second-line recurrent/metastatic SCCHN (N=192), the ORR was 18%, and the median duration of response was NR (range, 2+ to 30+ months [55, 56]. TRAEs occurred in 64% of patients, with the most common TRAEs being fatigue (22%), hypothyroidism (10%), rash (9%), pruritis, and appetite decrease (8% each).
A phase 2 study of nivolumab in patients with metastatic RCC (N= 168) showed an ORR of >20% across all doses, with a large proportion of responders (40%) experiencing an ongoing response at 24 months [57]. Most patients (73%) experienced a TRAE including fatigue (22%-35% of patients across dose groups), nausea (10%-13% across dose groups), and pruritis (9%-1 1% across dose groups). In a head-to- head phase 3 study, the median OS was 25.0 months (95% CI, 21.8-NR months) for nivolumab vs. 19.6 months (95% CI, 17.6-23.1 months) for the mechanistic target of rapamycin inhibitor, everolimus. The ORR was 25% Vs 5% (odds ratio, 5.98) [58]. Nivolumab is now recommended by the National Comprehensive Cancer Network for second-line treatment of RCC (NCCN, Kidney Cancer, Version 1.2021). TRAEs occurred in 79% of patients receiving nivolumab, with the most common TRAEs being fatigue (33%), nausea, and pruritis (14% each). Prior to the use of anti-PD-1/PD-L1 therapies in UC, the prognosis of patients receiving second-line chemotherapy was very poor. Pembrolizumab previously showed survival benefit VS chemotherapy in patients with advanced UC in a phase 3 study, with a median OS of 10.3 months (95% CI, 8.0-11.8 months) Vs 7.4 months (95% CI, 6.1-8.3 months). TRAEs occurred in 60.9% of patients receiving pembrolizumab; the most common TRAEs were pruritis (19.5%), fatigue (13.9%), and nausea (10.9%) [59]. Although the first-line SOC for patients with locally advanced or metastatic UC is platinum-based chemotherapy, it provides limited long-term benefits because the median progression- free survival is approximately 6 to 8 months and OS is approximately 8 to 15 months, likely due to development of chemoresistance [60, 61, 62]. In the phase 3 JAVELIN Bladder 100 trial, avelumab first-line maintenance significantly prolonged OS vs best supportive care alone (21.4 months [95% CI, 18.9-26.1 months] vs 14.3 months [95% CI, 12.9- 17.9 months]; HR, 0.69) in patients with UC that had not progressed on first-line chemotherapy [63]. TRAEs were reported in 98.0% of patients receiving avelumab, the most common of which were fatigue (17.7%), pruritis, and urinary tract infection (17.2% each).
Mechanism of Dual Targeting PD-L1 and TGF-β
Despite improvements in treatment outcomes across a variety of tumor types, as of 2019, the median ORR for PD-1/PD-L1 monotherapy in solid tumors is approximately 20%, indicating that a significant unmet need remains [64]. Although durable antitumor responses can be achieved with approved PD-1/PD-L1 inhibitors, some patients never respond to anti-PD-1/PD-L1 therapy (primary refractory), while others develop resistance after receiving anti-PD-1/ PD-L1 therapy (acquired resistance) [65]. There are three main phenotypes associated with anti-PD-1/PD-L1 resistance: a TME lacking lymphocytes (“immune desert”), a TME in which lymphocytes are physically excluded from tumor cells (“immune excluded”), and a TME in which lymphocytes infiltrate the tumor tissue (“inflamed”) but are inactivated through a negative feedback loop of PD-L1 signaling, resulting in T-cell exhaustion [66, 67].
TGF-β and PD-L1 are nonredundant pathways driving immunosuppression in the TME [9, 48]. TGF-β can promote PD-1/PD-L1 resistance by converting conventional T cells to immunosuppressive Treg cells and increasing the survival of myeloid progenitors that differentiate to potent MDSCs [25, 68]. Both of these processes result in increased expression of TGF-β, while MDSCs express PD-L1 and drive Treg cell differentiation [25, 68]. In a preclinical study, inhibition of TGF-β reduced the number of Treg cells, increased the number of effector T cells, and restored sensitivity to anti-PD-L1 therapy [69]. As we know CAFs serve as a barrier to T-cell infiltration of the tumor parenchyma. In the context of an immune-excluded phenotype, TGF-β promote activation of CAFs resulting limited efficacy of anti-PD-L1 therapy in bladder cancer [17]. Lastly, T-cell exhaustion is a phenomenon in which tumors exhibit robust immune infiltration within the TME, such as in the inflamed phenotype, but are ineffective in controlling tumor growth. PD-L1 binding of the receptor PD-1 on immune cells can promote negative feedback to T cells. In addition, T cells express other checkpoint molecules, such as TIGIT, LAG-3, and TIM3, to limit their cytotoxic activity [66].
Given the importance of the TGF-β and PD-L1 pathways in the development of cancer, the simultaneous inhibition of these pathways may potentially enhance the antitumor activity when each pathway is targeted alone. A recent analysis of plasma levels of soluble TGF-β and PD-L1 in 90 patients treated with first-line chemotherapy found that there was a positive correlation between TGF-β and PD- L1 at baseline and after treatment and that an increase in soluble TGF-β levels following chemotherapy was associated with worse prognosis [70]. In addition, in a biomarker analysis of pretreatment tumor samples from the phase 2 IMvigor210 trial, high expression of TGF-β was associated with lack of response to PD-L1 blockade with atezolizumab [17]. Inhibition of TGF-β may thus remove a barrier in solid tumors to immune checkpoint inhibitor-based therapy [71]. Furthermore, in PD-L1-positive tumors, the TGF-β is concentrated in the TME. Thus, a single molecule targeting PD-L1 and TGF-β may ensure suppression of TGF-β signaling in the TME compared with independent dual combination therapies. A preclinical study evaluated the anticancer efficacy of 2 bifunctional molecules: one targeting TGF-β and PD-L1 and the other targeting TGF-β and cytotoxic T-lymphocyte associated-protein 4 (CTLA-4). Both bifunctional molecules were superior to their parent immune checkpoint inhibitors (atezolizumab and ipilimumab, respectively) used alone or in combination with the TGF βRII domain in causing tumor regression in murine models [72]. An additional bispecific antibody for TGF-β and PD-L1, YM101, was recently described in a preclinical study. YM101 demonstrated the ability to bind all three isoforms of TGF-β, and its antitumor activity was better than the combination of anti-TGF-β and anti-PD-L1 treatments in mouse tumor models [73].
Bintrafusp alfa, also known as M7824, is a first-in-class bifunctional fusion protein composed of the extracellular domain of the TGF-βRII receptor to function as a TGF-β “trap” fused to a human immunoglobulin G1 monoclonal antibody blocking PD-L1. In a preclinical study, it was demonstrated that simultaneous inhibition of both pathways using a bifunctional approach resulted in superior antitumor activity compared with either the TGF-β “trap” or anti- PD-L1 antibody [74]. In the absence of a specific anti-PD-L1 moiety, the TGF-β trap reduced the plasma levels of TGF-β1 but did not decrease the TGF-β-dependent signaling in the TME [75], indicating the importance of the bifunctional nature of bintrafusp alfa to ensure that suppression of TGF-β occurs only in the TME. In preclinical studies, bintrafusp alfa sequestered all three isoforms of TGF-β in the TME and bound efficiently and specifically to PD-L1 both in vitro and in vivo. This resulted in superior tumor regression compared with a TGF-β “trap”, an anti-PD-L1 antibody, or combination of the both [74]. It could be an avidity effect that the isolated TGF-βRII ectodomain shows high binding affinity toward TGF-β1 and TGF-β3 but not TGF-β2, and yet bintrafusp alfa neutralizes all three circulating isoforms in both mice and humans [74, 76]. This is because the obligatory dimeric conformation of the receptors at the C terminus of the antibody allows bivalent binding of the TGF-β homodimer. A radiolabeling study showed that bintrafusp alfa localized to the PD-L1-rich tissues such as spleen and lung, indicating bintrafusp alfa localizes to the PD-L1-rich TME in vivo [77]. Preclinical data showed cancer fibrosis was reduced by bintrafusp alfa treatment, which may result in (1) enhanced immune cell access to the tumor, (2) restored drug access to the tumor, and (3) reduced metastatic potential of the tumor [74, 75, 78]. Bintrafusp alfa has the potential to inhibit angiogenesis through suppression of TGF-β activity via stromal modulation and may restore normal vascular homeostasis, thereby enhancing drug delivery and T-cell infiltration into the TME [79, 80, 81]. Likewise, bintrafusp alfa may reduce the expression of VEGF and subsequent angiogenesis by inhibiting TGF-β signaling [69].
Given the effect of radiation therapy could increase TGF-β activation and immunogenicity via antigen release, bintrafusp alfa may be a suitable combination partner for radiation therapy by counteracting TGF-β signaling, increasing infiltration of CD8+ T cells, and enhancing the abscopal effect [74, 82, 83, 84]. In addition, bintrafusp alfa may reduce radiation-induced fibrosis, which has been linked to treatment resistance [74, 85]. Similarly, preclinical data support synergistic effects with the combination of bintrafusp alfa and chemotherapy because TGF-β inhibition may normalize the extracellular matrix, improve drug delivery and resistance via its effects on fibrosis, EMT, and angiogenesis as well as elimination of chemotherapy-resistant cancer stem-like cells [74, 86]. Simultaneous targeting of two nonredundant immunosuppressive pathways may result in superior antitumor activity (Figure 1).
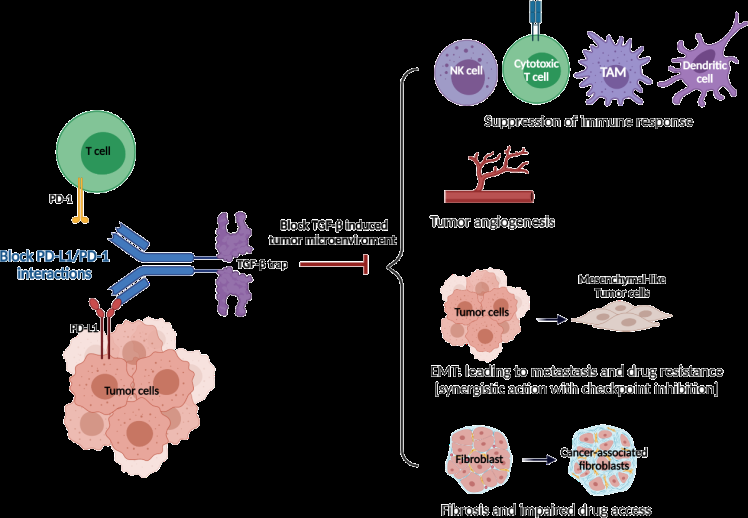
Source: Cancer cells escape from immune surveillance by activating immune checkpoint pathways such as PD-1/PD- L1 pathway. PD-1 is usually transiently expressed on T cells during priming and expansion. The binding of PD-1 to PD-L1 inhibits the function of T cells. The PD-1/PD-L1 axis is not only an important feedback loop of immune homeostasis but also participates in tumor immune evasion. Transforming growth factor-beta (TGF-β) is usually overexpressed in advanced tumors and related to poor prognoses. TGF-β promotes distant metastasis, drug resistance, and immune escape in advanced cancer. In the TME with hyperactive TGF-β signaling, the effect of anti-PD-1/PD-L1 therapy is limited. Figure 1: Rationale of dual targeting PD-L1 and TGF-β.
Current Treatment Strategy of Dual Targeting PD-L1 and TGF-β
To date, there are two single-molecule TGF-β/PD-L1 inhibitors in clinical development: bintrafusp alfa and SHR- 1701. Bintrafusp alfa is the most developed single-molecule therapy, with multiple phase 2 and 3 trials in biliary tract cancer, NSCLC, cervical cancer, and other solid tumors [74, 75, 76, 84, 87, 88]. SHR-1701, another TGF-βRII/PD-L1 bifunctional fusion protein, is currently being evaluated in several phase 1/2 studies and one phase 3 study in patients with locally advanced/metastatic solid tumors.
Results from two phase 1 studies (NCT02517398 and NCT02699515) of more than 670 patients treated with bintrafusp alfa demonstrated promising clinical activity in various expansion cohorts of advanced solid tumors [76, 88, 89, 90, 91, 92, 93, 94, 95]. Durable responses of more than six months were reported in multiple advanced solid tumor types, and favorable response rates were observed when compared with historical data for anti-PD-1/PD-L1 therapies in the same tumor type, although no head-to-head studies have been conducted. Additionally, the safety profile appeared manageable and consistent with co-localized, simultaneous inhibition of the TGF-β and PD-L1 pathways. The most common TRAEs were rash maculopapular, pruritus, rash, asthenia, and hypothyroidism. TRAEs leading to discontinuation of bintrafusp alfa occurred at a rate of 6% to 20%. Skin lesions, including those that have been observed with fresolimumab, were observed at a rate of 3% to 13.3% across 8 expansion cohorts.
However, bintrafusp alfa encountered triple failure in clinical studies. In January 20, 2021, Merck announced bintrafusp alfa failed to show any improvement over Merck & Co/MSD’s checkpoint inhibitor Keytruda (pembrolizumab) in previously-untreated patients with PD-L1-positive non- small cell lung cancer (NSCLC). In March 16, 2021, Merck announced topline data from the Phase 2 INTR@PID BTC 047 study evaluating bintrafusp alfa as a monotherapy in the second-line treatment of patients with locally advanced or metastatic biliary tract cancer (BTC). Bintrafusp alfa missed the mark to meet the pre-defined threshold that would have enabled regulatory filing for BTC in the second line setting. In August 23, 2021, the Phase 2 INTR@PID BTC 055 study evaluating bintrafusp alfa with gemcitabine plus cisplatin in the first-line treatment of patients with locally advanced or metastatic biliary tract cancer (BTC) was abandoned, as the study is unlikely to achieve the primary objective of overall survival.
Another clinical stage dual targeting agent SHR-1701 presented relieved data. In a phase 1 dose-escalation and -expansion study (NCT03710265), no dose-limiting toxicities were observed, and the maximum tolerated dose was not reached for patients (N=49) with advanced solid tumors receiving SHR-1701; in patients who were evaluable for efficacy (n=45), the ORR was 17 .8% (95% CI, 8.0%-32. 1%), with 7 of 8 responses ongoing. The most common TRAEs (incidence >15%) were increased alanine aminotransferase/ aspartate aminotransferase, anemia, hypothyroidism, and increased bilirubin/conjugated bilirubin [96]. In an expansion cohort of a phase 1b study (NCT03774979), patients with epidermal growth factor-positive NSCLC (N=27; 24 were evaluable for efficacy) receiving SHR-1701 had an ORR of 16.7% (95% CI, 4.7%-37 .4%). Grade 3 TRAEs occurred in 7.4% of patients, including anemia, hypokalemia, and asthenia (3.7% each) [97].
Though bintrafusp alfa encountered difficulties in 2021, dual targeting TGF-β and PD-L1 therapy still demonstrated noticeable benefits and clear rationale in tumor inhibition. Here we discuss two accessible strategies to maximum the efficacy of dual targeting TGF-β and PD-L1 agents: choosing the proper indication and combination regimen with other synergistic therapy.
Further Cancer Indications
Triple Negative Breast Cancer: Breast cancer is the first most frequent cancer in women that its incidence has shown an increase over the last decade. The rate of 5-year metastasis or recurrence for such cancer is very high (1/3 patients) [98]. Estrogen receptor-positive (ER+)/human epidermal growth factor receptor 2-negative (HER2) subtype are the most common form of breast malignancy, which represents about 60–65% of all breast cancer cases [99]. Triple negative breast cancer (TNBC) represents the most aggressive subtype [100]. TNBC patients do not express progesterone receptor, estrogen receptor or HER2. Chemotherapy is the first-line treatment for metastatic TNBC, but the outcomes are not acceptable, requiring new therapeutic approaches [101]. Although screening mammography is important for early diagnosis of breast cancer, about 20–30% of patients are not detected by this way [102], and that the ER+ subtype may recur over 20 years from the initial diagnosis. The resistance generally occurs in patients with a metastatic disease [103]. Anti-tumor activity of the anti-PD-1/PD-L1 monotherapy has been studied in a number of patients with advanced cold cancers. Adams and colleagues administered pembrolizumab (200 mg) in the two groups of metastatic TNBC patients: subjects with previous history of systemic therapy [104], and cases without previous history of standard chemotherapy [101]. The duration of response at data cut- off was not reached in the first group, whereas the median response duration of 10.4 months was reached in the second report. The objective response rate (ORR), disease control rate (DCR), median progression-free survival (PFS), and median overall survival (OS) for patients in the first group were 5.7%, 9.5%, 2 months, and 9 months, while for patients in the second report were 21.4%, 23.8%, 2.1 months, and 18 months. The authors also noticed that despite showing lower ORR in comparison with the single agent chemotherapy, pembrolizumab treatment prevented common toxicities related to the chemotherapy regimen, and that responses to the anti-PD-1 were somewhat durable. Comparing the results from the two reports by Adams and colleagues sent an important message. In patients with advanced TNBC who are suitable for immunotherapy, the responses are more potent when they had no previous history of the systemic anti-cancer therapy. An interpretation that could be made for such differences are that patients with previous history of systemic therapy possibly developed mechanisms of resistance, rendering them lower responsive to the immunotherapy than the subjects without previous history of systemic therapy. A point to consider, however, is the sharp difference between the number of patients evaluated for each study (170 cases for the first report vs. 84 patients for the second report), which may influence the outcomes. To support the idea, Emens and colleagues compared the outcomes of ORR in patients with metastatic TNBC receiving azetolizumab, and they noticed the higher ORR (24%) for subjects with the first-line azetolizumab, compared to that of second line or third line (ORR: 6%) [105].
TGF-β1 has an important activity in breast cancer stem cells, as they express TGF-β1 and the TGF-β1 receptor exponentially [106]. TGF-β inhibitors can inhibit the growth and multiplication of chemotherapy-resistant tumor- initiating cells (TIC) in vivo, forming the basis for combinatorial chemotherapy for patients suffering from TNBC. TGF-β stimulates an epithelial-to-mesenchymal transition (EMT) within mammary cells, leading to an exhibition of tumor- like properties. It is possible to reverse EMT via TGF-βR1/2 inhibitors while stimulating mesenchymal-to-epithelial (MET) differentiation inside mammary epithelial cells [107]. TGF-β is frequently found overexpressed in the TNBC tumor microenvironment, especially in tumor cells, or by tumor-associated immune and stromal cells. These cells also activate SMAD2/3 and SMAD4, leading to metastasis and angiogenesis. This indicates that the TGF-β inhibitors play an important role in patients with metastasis [107].
Considering the poor prognosis of current treatment strategy, as well as the relatively clear benefits of PD-1/PD- L1 and TGF-β signaling blocking in anti-tumor treatment, Merck’s pipeline also lists ongoing phase 2 trials of HMGA2- positive triple-negative breast cancer (TNBC). The upcoming results may hand over more cogent evidence of dual targeting PD-1/PD-L1 and TGF-β signaling.
Cervical Cancer: Cervical carcinoma is one of the most common types of cancer worldwide and one of the leading causes of death from cancer in women. High risk human papillomaviruses (HPV) are known to play an important etiological role in cervical cancer development, but only a small part of infected women develop cervical cancer, as most infections with HPV regress spontaneously [108]. The presence of HPV infection has been detected in more than 90% of cervical cancers. HPV persistent infection is a prerequisite for the development of precancerous cervical lesions, termed cervical intraepithelial neoplasia, and cancer [109, 110]. HPV infection has been closely associated with increased TGF-β expression in a variety of solid tumors, including cervical cancer. Several studies showed an important role for TGF-β signaling pathway in cervical oncogenesis [111]. A particularly interesting biological characteristic associated with malignant progression of cervical epithelial cells is their progressive loss of responsiveness to TGF-β [112, 113]. An array study showed that TGF-β was amplified and overexpressed more than 2-fold in the cervical cancer group relative to its transcription and expression in the normal group [114].
The accelerated FDA approval of pembrolizumab validated the efficacy of anti-PD-1/PD-L1 therapy for patients with recurrent/metastatic cervical cancer. However, the objective response rate with pembrolizumab was only 14.3% in patients with PD-L1-expressing tumors [115]. To meet the clinical needs of the rest of 80% patients, clinical studies evaluating bintrafusp alfa for the treatment of patients with cervical cancer were launched. In ESMO congress 2021, Strauss et al. disclosed data from patients with immune checkpoint inhibitor–naive, recurrent/metastatic cervical cancer treated with bintrafusp alfa in phase 1 (INTR@PID 001; NCT02517398) and phase 2 (study 012; NCT03427411) studies, announced bintrafusp alfa had a manageable safety profile and demonstrating encourage clinical activity with an ORR of 28.0% [116, 117].
KRAS Mutated NSCLC: KRAS, one of the earliest oncogenic drivers, are mainly found in lung, pancreatic and colon cancers. KRAS mutations account for approximately 30% of lung adenocarcinomas in Western countries and for 10– 15% of cases in Asia [118]. The KRASG12C mutation is highly prevalent in patients suffering from lung adenocarcinoma (13% of total lung adenocarcinoma) and account for >50% of all KRAS mutant cases [119]. According to its structural and biochemical obstacles, effective KRAS-targeted therapies still remain elusive. KRAS mutant lung cancers have worse outcomes in both early stage and advanced metastatic settings, illustrating the critical need for novel agents targeting KRAS-driven NSCLC. Numerous therapeutic strategies have been developed including targeting KRAS membrane associations, synthetic lethality partners, blockage of downstream signaling cascades, targeting metabolic reprogramming, direct targeting of KRAS and immunotherapy.
PD-L1 expression is the only FDA-approved biomarker for these therapies in patients with lung adenocarcinoma, but its sensitivity is modest. Studies on the predictive importance of KRAS mutations for the efficacy of immune checkpoint inhibitors have not uniformly provided positive results. Mutant KRAS was associated with increase of tumor infiltrating lymphocytes, PD-L1 expression and tumor mutational burden [120, 121]. However, while Gianoncelli, et al. observed no significant differences between KRAS+ and non-KRAS NSCLC patients in terms of progression- free or overall survival [122], Kauffmann-Guerrero et al. reported a positive outcome for KRAS mutations in response to immune checkpoint inhibitors [123]. Molecular diversity within KRAS+ NSCLC patients offers an attractive biological explanation for such discrepancy [124]. In particular, co- alterations in STK11 and p53, both associated with high tumor mutational burden, profoundly influence the tumor- immune contexture [125]. KRAS/STK11 NSCLC (25% of KRAS+ NSCLC) lacked tumor-infiltrating lymphocytes and expressed reduced-levels of immune markers and PD- L1. Inactivating somatic mutations in STK11 have thus emerged as a major genomic driver of primary resistance to immune checkpoint inhibitors [126]. Such negative impact extends to tumors with high PD-L1 expression and is further associated with primary resistance to combined PD-L1/ CTLA-4 blockage in NSCLC [127]. Simultaneous therapies that reverse the T cell suppressive tumor microenvironment in KRAS/STK11 NSCLC would be required for a potent response. PD-1 targeting antibodies were ineffective against Stk11/Lkb1-deficient tumors. In contrast, treating Stk11/ Lkb1-deficient mice with an IL-6 neutralizing antibody or a neutrophil-depleting antibody yielded therapeutic benefits associated with reduced neutrophil accumulation and proinflammatory cytokine expression [128]. Similarly, anti- angiogenics and epigenetic modifiers may also needed for the treatment of KRAS/STK11 NSCLC. In addition, mutations in phosphatase and tensin homolog (PTEN) and KEAP1 have been considered as potential drivers of de novo resistance to immune checkpoint inhibition in NSCLC [127, 129]. Co- mutation in KEAP1 was also associated with shorter duration of initial platinum-based chemotherapy and shorter overall survival from start of immune therapy in KRAS+ advanced NSCLC [129]. In contrast, KRAS/p53 co-mutated NSCLC was characterized by an inflammatory response, immune-editing and expression of co-stimulatory and co-inhibitor molecules including PD-L1 [126, 130]. These observations indicate that the cohort may be particularly susceptible to anti-immune checkpoint therapies. Indeed, KRAS/p53 co-mutations were associated with potent treatment response and improved progression-free and overall survival [126]. Taken together, co-occurring somatic genomic alterations in KRAS+ NSCLC represent independent predictors for sensitivity to anti- PD-1/PD-L1 therapies. Treatment needs to be individualized and may require the use of rational combinations (e.g., immunotherapy plus conventional therapies targeting RAS downstream cascade or cell cycle inhibitors) for a durable therapy response in KRAS+ NSCLC [131]. In immune- competent mice, the KRASG12C inhibitor AMG 510 resulted in a pro-inflammatory tumor microenvironment and produced durable cures in combination with immune checkpoint inhibitors [132]. Several clinical trials are ongoing. A phase I study is evaluating the combination of the CDK4/6 inhibitor abemaciclib with anti-PD-1 antibody pembrolizumab for KRAS mutant and PD-L1+ patients with stage IV NSCLC (NCT02779751). A second trial is recruiting patients to determine the side effects and best dose of pembrolizumab plus the MEK inhibitor trametinib for recurrent KRAS- mutated NSCLC that is metastatic, unresectable or locally advanced (NCT03299088, NCT03225664). Another phase I study focuses on the safety and tolerability of pembrolizumab infusion in combination with a KRAS vaccine mRNA-5671/ V941 in patients with KRAS mutant, advanced or metastatic NSCLC (NCT03948763). An open-label phase II study aims to assess the safety and efficacy of anti-PD-1 antibody SHR- 1210 plus VEGF receptor-2 inhibitor apatinib mesylate versus metabolite inhibitor pemetrexed and alkylating agent carboplatin in adult patients with KRAS+ stage IV NSCLC (NCT03777124). Another phase II trial is underway to determine the effects of combining pembrolizumab with anti-VEGF receptor ramucirumab and docetaxel in treating NSCLC patients who failed to respond to treatments with platinum-based chemotherapy administered concurrently or sequentially with anti-PD-1/PD-L1 immunotherapy (NCT04340882).
TGF-β1-induced activation of EMT mediates tumor progression by enhancing invasion and metastasis in several cancers, including NSCLC [133]. Recently, Pan, et al. [134] showed that KRAS-mediated upregulation of PD-L1 promotes immune escape via TGF-β/EMT signaling pathway in KRAS- mutant NSCLC. They found that pembrolizumab activated the anti-tumor activity and decreased tumor growth with KRAS (G12V) mutation. Furthermore, in vivo assay revealed that pembrolizumab reversed EMT in KRAS-mutated tumors. While single agent PD-1/PD-L1 blocker presented limited efficacy in clinical stage, dual targeting TGF-β and PD-1/PD- L1 signaling is an attractive path.
Combination Therapy of PD-L1/TGF-β with Two or more Agents
Combination Treatment with Radiotherapy
Radiotherapy (RT) is one of the standard treatments for some malignant tumors, while the long-term radiation toxicity in normal tissue is commonly observed. Radiation- induced fibrosis (RIF) may have negative effects on aesthetic and functional prognosis and even life prognosis of cancer survivors [135].
The essential multi-step process of RIF includes release of reactive oxygen species (ROS), microvascular injury, recruitment of inflammatory cells, and activation of myofibroblasts [136]. SMAD-regulated connective tissue growth factor (CTGF) expression-mediated by TGF-β1 is believed to be the main signaling pathway involved in RIF. Excess production of ROS after radiation exposure can oxidize cysteine residues and change the conformation of the latency-associated peptide (LAP), disrupting the noncovalent bonds between LAP and TGF-β1 [137]. This allows bioactive TGF-β1 release from the latent complex and makes TGF-β1 accessible to its receptors on the cell surface, initiating an intracellular signaling cascade [138]. Activation of the TGF-β signaling pathway by ROS mechanistically promotes sustained high levels of DNA methyltransferases (DNMTs), histone deacetylases (HDACs), methyltransferases, and demethylases in different fibrotic models, and their upregulation can lead to downregulation of antifibrotic genes that prevent development of fibrosis [139, 140, 141]. TGF-β1 has been acknowledged as the master switch in the development of fibrosis, driving quiescent fibroblast proliferation from resident stromal fibroblasts and bone marrow progenitors [142]. TGF-β1 affects fibrogenic function through various signaling pathways, with which receptor-regulated SMAD signaling is referred as the main axis. TGF-β1 initiates its cellular response by binding to TGF-β1 receptor phosphorylates, i.e., Smads (Smad2 and Smad3). Subsequently, activated SMADs assemble with Smad4 and translocate into the nucleus where the SMAD complex interacts with canonical SMAD-binding elements of target genes to activate a profibrotic program of transcription [143]. CTGF, a typical member of the cellular communication network (CCN) matricellular protein family, has been considered as a downstream mediator of TGF-β1-induced activation of fibroblasts. A TGF-β1 response element was located in the CTGF promoter, indicating that CTGF is likely to be a fibroblast-selective effector of profibrotic effects of TGF-β1 [144]. CTGF binds to yet uncharacterized plasma membrane receptors and activates downstream fibrogenic signaling cascades [145]. Furthermore, the induction of CTGF can also act independently of TGF-β1/SMAD signaling through the activation of the Rho/ROCK pathway. Moreover, TGF-β1/SMAD and Rho/ROCK pathways and other signaling cascades such as PDGF/PDGFR have been associated with the activation of myofibroblast [146] (Figure 2).
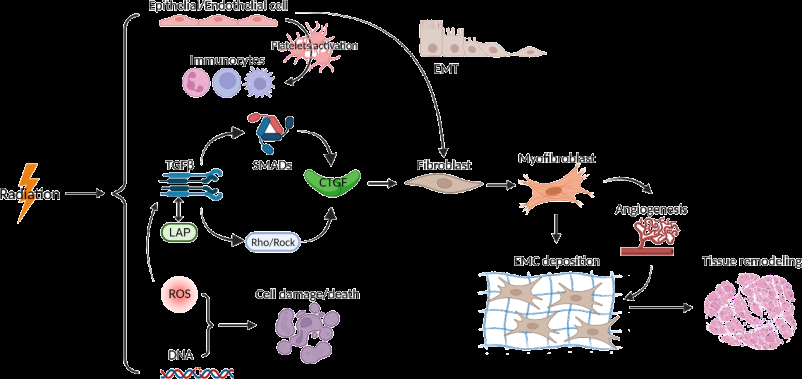
Source: Radiation induces the release of reactive oxygen species (ROS), microvascular injury, recruitment of inflammatory cells, and activation of myofibroblasts. ROS/Rock, resulting the expression of connective tissue growth factor (CTGF) expression. CTGF binds to yet uncharacterized plasma membrane receptors and activates downstream fibrogenic signaling cascades. Figure 2: Pathogenic mechanism of radiation-induced fibrosis.
Moreover, several preclinical studies have reported that irradiation induced PD-L1 expression [147, 148, 149]. Therefore irradiation interferes with the effector functions of interacting T cells. Preclinical studies demonstrate that radiotherapy induces PD-L1 upregulation by 4 pathways: (1) IFN-γ signaling, (2) EGFR pathway, (3) DNA damage signaling pathway, and (4) cGAS-STING pathway.
The IFN-dependent pathway is canonical pathway for PD-L1 expression. Although both type I and II IFNs can upregulate PD-L1, IFNγ induction of PD-L1 is stronger and more persistent via JAK–STAT–IRF pathway [150, 151, 152]. Dovedi et al. analyzed PD-L1 upregulation by X-ray RT using tumor-bearing mice [147]. They found that PD-L1 upregulated with a peak of 3 days after the last prescription of fractionated RT, which requires IFNγ production by CD8- positive T cells. Conversely, they also reported that in vitro single fractions of 2.5–10 Gy for the same murine cancer cell lines did not effectively induce PD-L1 expression, suggesting the importance of IFNγ in PD-L1 upregulation after X-ray irradiation. In addition, because type I IFN induces PD- L1 expression, cGAS–STING pathway is also an important upstream signal for PD-L1 expression. Recently, type I IFN induction via cGAS–STING pathway was shown to be induced after 24 Gy in three fractions of murine mammary tumor cell-bearing mice [153]. The IL-6/JAK/STAT pathway is also involved in PD-L1 upregulation in human esophageal cancer cells after irradiation [148]. Additionally, EGFR signaling after irradiation contributes to PD-L1 expression via the IL-6/JAK/STAT3 pathway [154, 155]. This PD- L1 expression was mediated by irradiation-dependent phosphorylation of EGFR (Y1173) and JAK2 (Y1007, Y1008) and was suppressed by inhibiting JAK2 phosphorylation, supporting the importance of EGFR–JAK signaling in PD-L1 expression after irradiation. In addition, preclinical studies have revealed that the response to DNA damages, such as DNA double-strand breaks, DNA single-strand breaks and base damage, upregulated PD-L1 expression in cancer cells via ATM/ATR/Chk1 kinase activation [156, 157, 158]. These data were further supported by experiments with mouse tumor models using a specific ATR inhibitor where RT-induced PD- L1 upregulation was significantly suppressed, resulting in the attenuation of RT-induced CD8-positive T cell exhaustion and cancer cells sensitized to the cytotoxicity of CD8-positive T cells [159, 160]. These studies suggest that the ATR/Chk1 activity followed by the activation of STAT–IRF pathway, rather than DNA damage per se, is a central factor that affects PD-L1 upregulation after irradiation.
These studies suggested that combination of ICIs with TGF-β blockade may mitigate RT-induced fibrosis, while ICIs to eradicate immune-cold tumors and TGF-β sequestration reduce overlapping toxicity. Lan et al found bintrafusp alfa reduces α-smooth muscle actin (α-SMA), a marker of CAF activation, and collagen deposition, as measured by picrosirius red staining, and increases CD8+ TILs in the EMT- 6 model [74]. Consistently, in the 4T1 model, BA significantly reduced stromal α-SMA expression versus isotype control. Although RT significantly increased α-SMA levels [85], consistent with RT activating CAFs, bintrafusp alfa was able to reverse RT-induced α-SMA. RT monotherapy increased tumor collagen deposition, resulting in tumors with thicker, more densely arranged collagen fibrils compared with isotype control or bintrafusp alfa-treated tumors. bintrafusp alfa reversed RT-induced collagen deposition versus RT, possibly as a result of reduced CAF activity. A similar reversal in RT-induced collagen deposition with bintrafusp alfa+RT (BART) was observed by alternative staining. Taken together, these data suggest that BA may reverse RT-induced CAF activation and fibrosis [161].
Recently, Yan, et al. [160] evaluated the anti-tumor activity of bintrafusp alfa plus RT therapy in several murine tumor models lacking tumor infiltrating lymphocyte. In Pdx-1-Cre KPC model mimics features of human pancreatic ductal adenocarcinoma, which including fibrosis deposition and an inflamed TME, exclusion of infiltrating effector T cells, and poor antitumor activity with ICI therapy alone, BART decreased tumor burden and metabolic activity comparing bintrafusp alfa or RT monotherapies, the combination of RT and TGF-β trap (p = 0.0007) or RT plus anti-PD-L1 (p = 0.0136). In Lewis lung carcinoma model which tumors generally lack TILs and are unresponsive to anti-PD-L1 therapy, significant tumor growth inhibition (TGI) was induced by both bintrafusp alfa (42.6%) and RT (52.8%) monotherapies relative to isotype control at day 11, BART further enhanced TGI (84.5%) relative to bintrafusp alfa or RT alone, and significantly prolonged survival with median survival of 26.5 days relative to 14 days (p < 0.0001) of bintrafusp alfa treatment or 18 days (p < 0.0001) of RT. Orthotopic GL261 glioblastoma tumors growing in brains of syngeneic C57BL/6 mice are relative resistance to RT [162]. They had limited TILs and sensitivity to anti-PD1 monotherapy. Bintrafusp alfa monotherapy slightly prolonged median survival (29 days) and increased complete tumor regressions in 3 of 10 mice relative to isotype control (23.5 days, p = 0.1379; 0 of 10 mice), RT monotherapy significantly prolonged median survival (38 days) and increased tumor regressions (2 of 10 mice) relative to isotype control (16 days, p < 0.0001; 0 of 10 mice), while BART further prolonged survival relative to RT (p = 0.0482) and increased tumor regressions (7 of 10 mice) to the end of the study (day 100) [162, 163]. In 4T1 breast cancer tumor model which has poor tumor infiltration of immune cells and is relatively resistant to ICIs, BART further induced TGI (67.5%) relative to bintrafusp alpha (TGI 27.2%) or RT (48.2%; p < 0.0001) monotherapy and significantly prolonged median survival (18 days) relative to bintrafusp alpah (12 days, p < 0.0001) or RT (14.5 days, p = 0.0021).
Combination with Chemotherapy
As described above, TGF-β is considered a master regulator of EMT and has been found to promote a phenotypic transition in many cancer models. The addition of TGF-β1 to lung cancer cell lines in vitro resulted decreasing susceptibility to the cytotoxic effect of docetaxel, paclitaxel and gemcitabine [78]. Experiments conducted in vivo also demonstrated the ability of bintrafusp alfa to increase anti-tumor efficacy of chemotherapy in a murine model of colon cancer. In MC38 immunoresponsive colon cancer model, combination treatment with bintrafusp alfa and the chemotherapies oxaliplatin and 5-fluorouracil (Ox/5-FU) reduced tumor volume and tumor weight more effectively than M7824 monotherapy or Ox/5-FU. Combination treatment also increased the frequency of p15E-reactive, IFN-γ–producing CD8+ T cells compared with M7824 or Ox/5-FU therapy alone, neither of which had a significant effect relative to isotype control [74].
Combination with Vascular Endothelial Growth Factor (VEGF) Inhibitors
Targeting tumor angiogenesis is an effective strategy for the treatment of solid tumors. TGF-β and VEGF are the main regulators of tumor angiogenesis, which can limit the efficacy of cancer treatment and inhibit anti-tumor immune responses. Combination therapy consisting of immune checkpoint inhibitors and tumor angiogenesis inhibitors is becoming an effective treatment for solid cancer. Yeung, et al. tested the anti-tumor efficacy of the bintrafusp alfa and VEGF blocker combination therapy [164]. In the CT26 mouse syngeneic colon cancer model, the combination of bintrafusp alfa with levatinib, regorafenib, axitinib or nintedanib showed significantly higher anti-tumor efficacy than the respective monotherapy. In the 4T1 mouse breast cancer model, the combination of bintrafusp alfa with VEGF inhibitor levatinib or regorafenib was more effective than the respective monotherapy. The combination of bintrafusp alfa and pazopanib also showed excellent anti- tumor efficacy in the orthotopic Renca mouse tumor model. These data indicate that PD-L1/ TGF-β dual blocker may be a viable combination partner with a variety of angiogenesis inhibitors, and provide a theoretical basis for the continued development of these combination therapies to treat solid cancer.
Combination with ALK1 Blocker
Activin receptor-like kinase 1 (ALK1) is an endothelial cell-restricted receptor of the TGF-β family. In endothelial cells, TGF-β has been shown to signal via both the ubiquitously expressed type I receptor ALK5 and through the predominantly endothelial cell restricted receptor ALK1
[165]. Recent study uncovers synergistic effect between anti-ALK1 agent and anti-PD-1 antibody nivolumab. Hsu et al disclosed data from a trial of GT90001 (Clinical trial information: NCT03893695), a fully human anti-ALK1 mAb (IgG2) that may inhibit ALK1/TGF-β signaling and tumor angiogenesis, showing promising preliminary antitumor activities combined with nivolumab as second-line treatment for patients with metastatic hepatocellular carcinoma (HCC) [166]. An objective response assessed was observed in 7 patients (43.75%) out of 16 evaluable patients, the disease control rate (complete response + partial response + stable disease) was 56.2%, while nivolumab single agent demonstrated an ORR for HCC of 20% as a first-line treatment or 14% as a second-line treatment in CheckMate 040 trial [167].
Combination with Ataxia Telangiectasia Mutated (ATM) Inhibitor
As discussed above, dual targeting of TGF-β and PD- L1 may reverse RT-induced CAF activation and fibrosis, activate cold tumor and therefore prolong survival in several tumor models. Inhibition of ATM increases the cancer cell’s sensitivity to radiotherapy by blocking cellular pathways which facilitate the response to, and repair of, RT-induced DNA double strand breaks. Lazorchak, et al. tested the anti-tumor efficacy of a combination therapy consisting of bintrafusp alfa, RT and the ATM kinase inhibitor M4076 [168]. Results suggested this triple combination therapy increased anti-tumor efficacy and prolonged overall survival relative to the bintrafusp alfa plus RT or M4076 plus RT dual combination therapies in the immune therapy resistant, 4T1 murine syngeneic breast carcinoma model. Further study in the more immunogenic MC38 colon carcinoma model presented consistent results. The bintrafusp alfa/RT/ M4076 triple therapy showed a significantly greater tumor growth inhibition and long-term tumor free survival when compared with the bintrafusp alfa plus RT, M4076 plus RT, or anti-PD-L1/RT/M4076 treatment groups. These data in the MC38 model suggest that bintrafusp alfa may promote a superior anti-tumor immune response relative to that of anti-PD-L1 when in combination with RT and M4076, and together support the potential rational benefits to develop a triple combination therapy of TGF-β and PD-L1/ RT/ ATMi for the treatment of solid tumors.
Conclusion
Anti-PD1/PD-L1 treatment could sometime overcome the immunosuppressive TME. However, the TGF-β pathway also plays a critical role in promoting tumor growth in multiple ways, including EMT, fibrosis, angiogenesis, and immunosuppression. The immunosuppression by TGF-β in TME is mainly achieved by promoting the function of Treg cells, tumor-associated macrophages and MDSCs, resulting in the inhibition of the killing function of NK cells as well as CD8+ T cells. TGF-β also directly inhibits the function of CD8+ T cells and NK cells. Therefore, inhibiting both PD1/ PD-L1 and TGF-β pathways could dramatically improve the immunosuppressive TEM. The superiority of the combination therapy of these two pathways has been well demonstrated in preclinical and clinical-stage studies. The dual-target treatment has excellent potential in the back- line therapy of advanced solid tumors. It is also an attractive potential therapy for first- and second-line therapy in solid tumors. Importantly, combination therapy of PD-L1/ TGF-β dual inhibitors with radiotherapy, chemotherapy, VEGF inhibitor, ALK1 inhibitor or ATM inhibitor could provide further benefits for the treatment of solid tumors.
References
-
Ciardiello D, Elez E, Tabernero J, Seoane J (2020) Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Annals of Oncology 2020 31(10): 1336-1349.
-
Heldin CH, Moustakas (2016) A Signaling receptors for TGF-β family members. Cold Spring Harbor Perspectives in Biology 8(8): a022053.
-
Batlle E, Massague J (2019) Transforming growth factor-β signaling in immunity and cancer. Immunity 50(4): 924-940.
-
Bulk JVD, Miranda NFCCD, Dijke PT (2021) Therapeutic targeting of TGF-β in cancer: Hacking a master switch of immune suppression. Clinical Science 135(1): 35-52.
-
Li MO, Flavell RA (2008) Contextual regulation of inflammation: a duet by transforming growth factor-β and interleukin-10. Immunity 28(4): 468-476.
-
Seoane J, Gomis RR (2017) TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harbor perspectives in biology 9(12): a022277.
-
Akhurst RJ, Hata A (2012) Targeting the TGFβ signalling pathway in disease. Nature reviews Drug discovery 11(10): 790-811.
-
Colak S, Ten Dijke PT (2017) Targeting TGF-β signaling in cancer. Trends in cancer 3(1): 56-71.
-
Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, et al. (2014) TGF-β: duality of function between tumor prevention and carcinogenesis. JNCI: Journal of the National Cancer Institute 106(2): djt369.
-
Agajanian M, Runa F, Kelber JA (2015) Identification of a PEAK1/ZEB1 signaling axis during TGFβ/fibronectin- induced EMT in breast cancer. Biochemical and biophysical research communications 465(3): 606-612.
-
Calon A, Lonardo E, Llergo AB, Espinet E, Momblona XH, et al. (2015) Stromal gene expression defines poor- prognosis subtypes in colorectal cancer. Nature genetics 47(4): 320-329.
-
Heldin CH, Vanlandewijck M, Moustakas A (2012) Regulation of EMT by TGFβ in cancer. FEBS letters 586(14): 1959-1970.
-
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4): 704-715.
-
Derynck R, Turley SJ, Akhurst RJ (2021) TGFβ biology in cancer progression and immunotherapy. Nature Reviews Clinical Oncology 18(1): 9-34.
-
Du B, Shim JS (2016) Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21(7): 965.
-
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, et al. (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165(1): 35-44.
-
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, et al. (2018) TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554(7693): 544-548.
-
Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P (2009) Transforming growth factor‐beta 1 (TGF‐β1) induces angiogenesis through vascular endothelial growth factor (VEGF)‐mediated apoptosis. Journal of cellular physiology 219(2): 449-458.
-
Ucuzian AA, Gassman AA, East AT, Greisler HP (2010) Molecular mediators of angiogenesis. Journal of Burn Care & Research 31(1): 158.
-
Ansems M, Span PN (2020) The tumor microenvironment and radiotherapy response; a central role for cancer- associated fibroblasts. Clinical and translational radiation oncology 22: 90-97.
-
Ghahremanifard P, Chanda A, Bonni S, Bose P (2020) TGF-β Mediated Immune Evasion in Cancer-Spotlight on Cancer-Associated Fibroblasts. Cancers 12(12): 3650.
-
Calon A, Espinet E, Ponce SP, Tauriello DV, Iglesias M, et al. (2012) Dependency of colorectal cancer on a TGF-β- driven program in stromal cells for metastasis initiation. Cancer cell 22(5): 571-584.
-
Liu T, Han C, Wang S, Fang P, Ma Z, et al. (2019) Cancer- associated fibroblasts: an emerging target of anti-cancer immunotherapy. Journal of hematology & oncology 12(1): 86.
-
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ (2013) The immunosuppressive tumour network: myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138(2): 105-115.
-
Junca AG, Driscoll KE, Pellicciotta I, Du S, Lo CH, et al. (2019) Autocrine TGFβ is a survival factor for monocytes and drives immunosuppressive lineage commitment. Cancer immunology research 7(2): 306-320.
-
Roncarolo MG, Levings MK, Traversari C (2001) Differentiation of T regulatory cells by immature dendritic cells. The Journal of experimental medicine 193(2): F5-F10.
-
Teicher BA (2007) Transforming growth factor-β and the immune response to malignant disease. Clinical Cancer Research 13(21): 6247-6251.
-
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-β:“N1” versus “N2” TAN. Cancer cell 16(3): 183-194.
-
Khan SA, Joyce J, Tsuda T (2012) Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC Res Notes 5: 636.
-
Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, et al. (2012) Phase I trial of “bi-shRNAifurin/GMCSF DNA/ autologous tumor cell” vaccine (FANG) in advanced cancer. Mol Ther 20(3): 679-686.
-
Ghisoli M, Barve M, Mennel R, Lenarsky C, Horvath S, et al. (2016) Three-year follow up of GMCSF/bi-shRNAfurin DNA-transfected Autologous tumor immunotherapy (vigil) in metastatic advanced Ewing’s sarcoma. Mol Ther 24(8): 1478-1483.
-
Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, et al. (2020) Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med 12(536): eaay8456.
-
Elez E, Kocakova I, Hohler T, Martens U, Bokemeyer C, et al. (2015) Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial. Ann Oncol 26(1): 132-140.
-
Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, et al. (2014) Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PloS one 9(3): e90353.
-
Gregory RC, Greco R, Qu H, Malkova N, Levit M, et al. (2018) The anti-TGFβ neutralizing antibody, SAR439459, blocks the immunosuppressive effects of TGFβ and inhibits the growth of syngeneic tumors in combination with anti-PD1. Cancer Res 78(S13): 2790.
-
Bauer TM, Lin CC, Greil R, Goebeler ME, Kroenke HML, et al. (2021) Phase Ib study of the anti-TGF-β monoclonal antibody (mAb) NIS793 combined with spartalizumab (PDR001), a PD-1 inhibitor, in patients (pts) with advanced solid tumors. Journal of Clinical Oncology 39(S15): 2509.
-
Tremblay G, Gruosso T, Denis JF, Figueredo R, Koropatnick J, et al. (2020) AVID200, a first-in-class selective TGF- beta 1 and-beta 3 inhibitor, sensitizes tumors to immune checkpoint blockade therapies. Cancer Res 80(S16): 6710.
-
Ballester B, Milara J, Cortijo J (2019) Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int J Mol Sci 20(3): 593.
-
Yap TA, Lakhani NJ, Araujo DV, Rodon Ahnert J, Chandana SR, et al. (2020) AVID200, first-in-class TGF-beta 1 and 3 selective and potent inhibitor: Safety and biomarker results of a phase I monotherapy dose-escalation study in patients with advanced solid tumors. Journal of Clinical Oncology 38(S15): 3587-3587.
-
Anido J, Borderias SA, Junca GA, Rodon L, Folch G, et al. (2010) TGF-β receptor inhibitors target the CD44high/ Id1high glioma-initiating cell population in human glioblastoma. Cancer cell 18(6): 655-668.
-
Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, et al. (2018) Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer 6(1): 47.
-
Keedy VL, Bauer TM, Clarke JM, Hurwitz H, Baek I, et al. (2018) Association of TGF-β responsive signature with anti-tumor effect of vactosertib, a potent, oral TGF-β receptor type I (TGFBRI) inhibitor in patients with advanced solid tumors. Journal of Clinical Oncology 36(S15): 3031.
-
Azhar M, Schultz JEJ, Grupp I, Dorn IIGW, Meneton P, et al. (2003) Transforming growth factor beta in cardiovascular development and function. Cytokine growth Factor Rev 14(5): 391-407.
-
Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, et al. (2011) Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol pathol 39(6): 916-924.
-
Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, et al. (2015) Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 9: 4479-4499.
-
Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, et al. (2017) A phase 1 study of anti-TGFβ receptor type- II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer chemother pharmacol 79(4): 673-680.
-
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer–immune set point. Nature 541(7637): 321-330.
-
Ferris RL, Lenz HJ, Trotta AM, Foncillas GJ, Schulten J, et al. (2017) Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev 63: 48-60.
-
Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, et al. (2016) Programmed death-ligand 1 expression and response to the anti–programmed death 1 antibody Pembrolizumab in melanoma. J Clin Oncol 34(34): 4102- 4109.
-
Eissler N, Mao Y, Brodin D, Reutersward P, Svahn AH, et al. (2016) Regulation of myeloid cells by activated T cells determines the efficacy of PD-1 blockade. Oncoimmunology 5(12): e1232222.
-
Bluthgen MV, Besse B (2015) Second-line combination therapies in nonsmall cell lung cancer without known driver mutations. Eur Respir Rev 24(138): 582-593.
-
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, et al. (2015) Pembrolizumab for the treatment of non– small-cell lung cancer. N Engl J Med 372(21): 2018-2028.
-
Ascierto PA, Long GV, Robert C, Brady B, Dutriaux C, et al. (2019) Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with nivolumab therapy: JAMA Oncol 5(2): 187-194.
-
Robert C, Long GV, Brady B, Dutriaux C, Maio M, et al. (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372(4): 320-330.
-
Chow LQ, Haddad R, Gupta S, Mahipal A, Mehra R, et al. (2016) Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/ or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol 34(32): 3838-3845.
-
Mehra R, Seiwert TY, Gupta S, Weiss J, Gluck I, et al. (2018) Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long-term follow-up in KEYNOTE-012. Br J Cancer 119(2): 153-159.
-
Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, et al. (2015) Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. Journal of clinical oncology 33(13): 1430-1437.
-
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, et al. (2015) Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373(19): 1803-1813.
-
Bellmunt J, De Wit R, Vaughn DJ, Fradet Y, Lee J-L, et al. (2017) Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med 376(11): 1015-1026.
-
De Santis M, Bellmunt J, Mead G, Kerst JM, Leahy M, et al. (2012) Randomized Phase II/III Trial Assessing Gemcitabine/Carboplatin and Methotrexate/ Carboplatin/Vinblastine in Patients with Advanced Urothelial Cancer Who are Unfit for Cisplatin-Based Chemotherapy: EORTC Study 30986. Journal of Clinical Oncology 30(2): 191.
-
Dogliotti L, Cartenì G, Siena S, Bertetto O, Martoni A, et al. (2007) Gemcitabine plus Cisplatin Versus Gemcitabine plus Carboplatin as First-Line Chemotherapy in Advanced Transitional Cell Carcinoma of the Urothelium: Results of a Randomized Phase 2 Trial. Eur Urol 52(1): 134-141.
-
von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, et al. (2005) Long-Term Survival Results of a Randomized Trial Comparing Gemcitabine Plus Cisplatin, with Methotrexate, Vinblastine, Doxorubicin, Plus Cisplatin in Patients with Bladder Cancer. J Clin Oncol 23(21): 4602-4608.
-
Powles T, Park SH, Voog E, Caserta C, Valderrama BP, et al. (2020) Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. The New England Journal of Medicine 383(13): 1218-1230.
-
Schmidt EV (2019) Developing Combination Strategies Using PD-1 Checkpoint Inhibitors to Treat Cancer. Semin Immunopathol 41(1): 21-30.
-
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168(4): 707-723.
-
Liu D, Jenkins RW, Sullivan RJ (2019) Mechanisms of Resistance to Immune Checkpoint Blockade. Am J Clin Dermatol 20(1): 41-54.
-
Trujillo JA, Sweis RF, Bao R, Luke JJ (2018) T Cell– Inflamed Versus Non-T Cell–Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol Res 6(9): 990-1000.
-
Wan YY, Flavell RA (2007) ‘Yin–Yang’ Functions of Transforming Growth Factor‐Β and T Regulatory Cells in Immune Regulation. Immunol Rev 220(1): 199-213.
-
Courau T, Nehar-Belaid D, Florez L, Levacher B, Vazquez T, et al. (2016) TGF-Β and VEGF Cooperatively Control the Immunotolerant Tumor Environment and the Efficacy of Cancer Immunotherapies. JCI insight 1(9): e85974.
-
Kim JW, Lee KH, Kim JW, Suh KJ, Nam AR, et al. (2021) The Prognostic Role of Soluble Transforming Growth Factor‐Β and Its Correlation with Soluble Programmed Death‐Ligand 1 in Biliary Tract Cancer. Liver Int 41(2): 388-395.
-
Groeneveldt C, van Hall T, van der Burg SH, Ten Dijke P, van Montfoort N (2020) Immunotherapeutic Potential of TGF-Β Inhibition and Oncolytic Viruses. Trends in immunology 41(5): 406-420.
-
Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, et al. (2018) Bifunctional Immune Checkpoint-Targeted Antibody-Ligand Traps that Simultaneously Disable Tgfβ Enhance the Efficacy of Cancer Immunotherapy. Nature communications 9(1):1-14.
-
Yi M, Zhang J, Li A, Niu M, Yan Y, et al. (2021) The Construction, Expression, and Enhanced Anti-Tumor Activity of YM101: A Bispecific Antibody Simultaneously Targeting TGF-Β and PD-L1. J Hematol Oncol 14(1): 27.
-
Lan Y, Zhang D, Xu C, Hance KW, Marelli B, et al. (2018) Enhanced Preclinical Antitumor Activity of M7824, a Bifunctional Fusion Protein Simultaneously Targeting PD-L1 and TGF-Β. Sci Transl Med 10(424): eaan5488.
-
Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, et al. (2018) M7824, A Novel Bifunctional Anti-PD-L1/Tgfβ Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination with Vaccine. Oncoimmunology 7(5): e1426519.
-
Strauss J, Heery CR, Schlom J, Madan RA, Cao L (2018) Phase I Trial of M7824 (MSB0011359C), A Bifunctional Fusion Protein Targeting PD-L1 and Tgfβ, in Advanced Solid Tumors. Clin Cancer Res 24(6): 1287-1295.
-
Burvenich IJG, Goh YW, Guo N, Gan HK, Rigopoulos A, et al. (2021) Radiolabelling and Preclinical Characterization of 89 Zr-Df-Radiolabelled Bispecific Anti-PD-L1/TGF- Βrii Fusion Protein Bintrafusp Alfa. Eur J Nucl Med Mol Imaging 48(10): 3075-3088.
-
David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, et al. (2017) A Novel Bifunctional Anti-PD-L1/ TGF-Β Trap Fusion Protein (M7824) Efficiently Reverts Mesenchymalization of Human Lung Cancer Cells. Oncoimmunology 6(10): e1349589.
-
Cox TR, Erler JT (2014) Molecular Pathways: Connecting Fibrosis and Solid Tumor Metastasis. Clin Cancer Res 20(14): 3637-3643.
-
Eser PÖ, Jänne PA (2018) Tgfβ Pathway Inhibition in the Treatment of Non-Small Cell Lung Cancer. Pharmacol Ther 184: 112-130.
-
Jain RK (2013) Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J Clin Oncol 31(17): 2205.
-
Barcellos-Hoff M, Derynck R, Tsang M, Weatherbee J (1994) Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest 93(2): 892-899.
-
Demaria S, Bhardwaj N, McBride WH, Formenti SC (2005) Combining Radiotherapy And Immunotherapy: A Revived Partnership. Int J Radiat Oncol Biol Phys 63(3): 655-666.
-
Morillon Y, Smalley Rumfield C, Pellom ST, Sabzevari A, Roller NT, et al. (2020) The Use of a Humanized NSG-Β2m−/− Model for Investigation of Immune and Anti-Tumor Effects Mediated by the Bifunctional Immunotherapeutic Bintrafusp Alfa. Front Oncol 10: 549.
-
Barker HE, Paget JT, Khan AA, Harrington KJ (2015) The tumor microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 15(7): 409-425.
-
Chen J, Ding ZY, Li S, Liu S, Xiao C, et al. (2021) Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics 11(3): 1345.
-
Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, et al. (2020) Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: status of preclinical and clinical advances. J Immunother Cancer 8(1): e000433.
-
Paz AL, Kim TM, Vicente D, Felip E, Lee DH, et al. (2020) Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol 15(7): 1210-1222.
-
Cho BC, Daste A, Ravaud A, Salas S, Isambert N, et al. (2020) Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer 8(2): e000664.
-
Doi T, Fujiwara Y, Koyama T, Ikeda M, Helwig C, et al. (2020) Phase I Study of the Bifunctional Fusion Protein Bintrafusp Alfa in Asian Patients with Advanced Solid Tumors, Including a Hepatocellular Carcinoma Safety‐ Assessment Cohort. Oncologist 25(9): e1292-e1302.
-
Kang YK, Bang YJ, Kondo S, Chung HC, Muro K, et al. (2020) Safety and Tolerability of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Asian Patients with Pretreated Recurrent or Refractory Gastric Cancer. Clin Cancer Res 26(13): 3202-3210.
-
Khasraw M, Weller M, Lorente D, Kolibaba K, Lee CK, et al. (2021) Bintrafusp alfa (M7824), a bifunctional fusion protein targeting TGF-β and PD-L1: results from a phase I expansion cohort in patients with recurrent glioblastoma. Neuro-oncology Advances 3(1): vdab058.
-
Lin CC, Doi T, Muro K, Hou MM, Esaki T, et al. (2021) Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Patients with Esophageal Squamous Cell Carcinoma: Results from a Phase 1 Cohort in Asia. Target Oncol 16(4): 447-459.
-
Tan B, Khattak A, Felip E, Kelly K, Rich P, et al. (2021) Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Esophageal Adenocarcinoma: Results from a Phase 1 Cohort. Target Oncol 16(4): 435-446.
-
Yoo C, Oh DY, Choi HJ, Kudo M, Ueno M, et al. (2020) Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with pretreated biliary tract cancer. J Immunother Cancer 8(1): e000564.
-
Liu D, Gong J, Liu T, Li K, Yin X, et al. (2021) Phase 1 study of SHR-1701, a bifunctional fusion protein targeting PD- L1 and TGF-β, in patients with advanced solid tumors. In.: Wolters Kluwer Health 29(15).
-
Shi M, Chen J, Li K, Fang Y, Wen G, et al. (2021) SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-β, for advanced NSCLC with EGFR mutations: Data from a multicenter phase 1 study. In.: Wolters Kluwer Health 39(15).
-
Liang S, Niu L, Xu K, Wang X, Liang Y, (2017) Tumor cryoablation in combination with natural killer cells therapy and Herceptin in patients with HER2- overexpressing recurrent breast cancer. Mol Immunol 92: 45-53.
-
Rugo HS, Delord JP, Im SA, Ott PA, Piha PSA, et al. (2018) Safety and antitumor activity of pembrolizumab in patients with estrogen receptor–positive/human epidermal growth factor receptor 2–negative advanced breast cancer. Clin Cancer Res 24(12): 2804-2811.
-
Nguyen KT, Druhan LJ, Avalos BR, Zhai L, Rauova L, et al. (2020) CXCL12-CXCL4 heterodimerization prevents CXCL12-driven breast cancer cell migration. Cell Signal 66: 109488.
-
Adams S, Loi S, Toppmeyer D, Cescon D, De Laurentiis M, et al. (2019) Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol 30(3): 405-411.
-
Grassmann F, He W, Eriksson M, Gabrielson M, Hall P, et al. (2019) Interval breast cancer is associated with other types of tumors. Nat Commun 10(1): 1-9.
-
Buschhaus JM, Humphries BA, Eckley SS, Robison TH, Cutter AC, et al. (2020) Targeting disseminated estrogen-receptor-positive breast cancer cells in bone marrow. Oncogene 39(34): 5649-5662.
-
Adams S, Schmid P, Rugo H, Winer E, Loirat D, et al. (2019) Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann Oncol 30(3): 397-404.
-
Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, et al. (2019) Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: a phase 1 study. JAMA Oncol 5(1): 74-82.
-
Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, et al. (2015) Therapeutic targets of triple‐negative breast cancer: a review. Br J Pharmacol 172(17): 4228- 4237.
-
Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, et al. (2013) TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 123(3): 1348-1358.
-
Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, et al. (1999) Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 59(24): 61320- 6136.
-
Woodman CB, Collins SI, Young LS (2007) The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer 7(1): 11-22.
-
Zur Hausen H (2002) Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2(5): 342-350.
-
Allan S, Braiteh F, Aller EC, Cervantes A, Edenfield W, et al. (2019) P37 Phase 1 evaluation of bintrafusp alfa (M7824), a bifunctional fusion protein targeting TGF-β and PD-L1, in cervical cancer. International Journal of Gynecological Cancer 29(4): A72.
-
Torng PL, Chan WY, Lin CT, Huang SC (2003) Decreased expression of human papillomavirus E2 protein and transforming growth factor‐β1 in human cervical neoplasia as an early marker in carcinogenesis. J Surg Oncol 84(1): 17-23.
-
Kang SH, Won K, Chung HW, Jong HS, Song YS, et al. (1998) Genetic integrity of transforming growth factor β (TGF‐β) receptors in cervical carcinoma cell lines: loss of growth sensitivity but conserved transcriptional response to TGF‐β. International journal of cancer 77(4): 620-625.
-
Yan D, Yi S, Chiu WC, Qin LG, Kin WH, et al. (2017) Integrated analysis of chromosome copy number variation and gene expression in cervical carcinoma. Oncotarget 8(65): 108912-108922.
-
Strauss J, Gatti Mays M, Cho B, Hill A, Salas S, et al. (2021) 957O Long-term follow-up of patients (pts) with human papillomavirus (HPV)–associated malignancies treated with bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1. Annals of Oncology 32(5): S829-866.
-
Strauss J, Braiteh F, Calvo E, De Miguel M, Cervantes A, et al. (2021) Evaluation of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in cervical cancer: Data from phase 1 and phase 2 studies. Journal of Clinical Oncology 39(15).
-
Dearden S, Stevens J, Wu YL, Blowers D (2013) Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of oncology 24(9): 2371-2376.
-
Bar Sagi D, Knelson EH, Sequist LV (2020) A bright future for KRAS inhibitors. Nature Cancer 1(1): 25-27.
-
Schoenfeld AJ, Rizvi H, Bandlamudi C, Sauter JL, Travis WD, et al. (2020) Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Annals of Oncology 31(5): 599-608.
-
Lee CK, Man J, Lord S, Cooper W, Links M, et al. (2018) Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non–small cell lung carcinoma: a systematic review and meta-analysis. JAMA oncology 4(2): 210-216.
-
Gianoncelli L, Spitaleri G, Passaro A, Radice D, Fumagalli C, et al. (2020) Efficacy of anti-PD1/PD-L1 therapy (IO) in KRAS mutant non-small cell lung cancer patients: a retrospective analysis. Anticancer research 40(1): 427-433.
-
Guerrero DK, Tufman A, Kahnert K, Bollmann BA, Reu S, et al. (2020) Response to checkpoint inhibition in non-small cell lung cancer with molecular driver alterations. Oncology research and treatment 43(6): 289-298.
-
Skoulidis F, Heymach JV (2019) Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nature Reviews Cancer 19(9): 495-509.
-
Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, et al. (2015) Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer discovery 5(8): 860-877.
-
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, et al. (2018) STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer discovery 8(7): 822-835.
-
Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez Vega F, et al. (2018) Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer cell 33(5): 843-852.
-
Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, et al. (2016) STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer research 76(5): 999-1008.
-
Arbour KC, Jordan E, Kim HR, Dienstag J, Helena AY, et al. (2018) Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non–small cell lung cancer. Clinical Cancer Research 24(2): 334- 340.
-
Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, et al. (2017) Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clinical cancer research 23(12): 3012-3024.
-
Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz Ares L, et al. (2018) KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung cancer 124: 53-64.
-
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, et al. (2019) The clinical KRAS (G12C) inhibitor AMG 510 drives anti- tumour immunity. Nature 575(7781): 217-223.
-
Shi S, Zhao J, Wang J, Mi D, Ma Z (2017) HPIP silencing inhibits TGF-β1-induced EMT in lung cancer cells. International journal of molecular medicine 39(2): 479-483.
-
Pan LN, Ma YF, Li Z, Hu JA, Xu ZH (2021) KRAS G12V mutation upregulates PD‐L1 expression via TGF‐β/EMT signaling pathway in human non‐small‐cell lung cancer. Cell Biology International 45(4): 795-803.
-
Rosenthal DI, Lewin JS, Eisbruch A (2006) Prevention and treatment of dysphagia and aspiration after chemoradiation for head and neck cancer. Journal of clinical oncology 24(17): 2636-2643.
-
Wang B, Wei J, Meng L, Wang H, Qu C, et al. (2020) Advances in pathogenic mechanisms and management of radiation-induced fibrosis. Biomedicine & Pharmacotherapy 121: 109560.
-
Richter K, Kietzmann T (2016) Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell and tissue research 365(3): 591-605.
-
Biernacka A, Dobaczewski M, Frangogiannis NG (2011) TGF-β signaling in fibrosis. Growth factors 29(5): 196-202.
-
Pang M, Zhuang S (2010) Histone deacetylase: a potential therapeutic target for fibrotic disorders. Journal of Pharmacology and Experimental Therapeutics 335(2): 266-272.
-
Chen PJ, Huang C, Meng XM, Li J (2015) Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 116: 61-69.
-
Irifuku T, Doi S, Sasaki K, Doi T, Nakashima A, et al. (2016) Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney international 89(1): 147-157.
-
Yarnold J, Brotons MCV (2010) Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol 97(1): 149-161.
-
Gauldie J, Bonniaud P, Sime P, Ask K, Kolb M (2007) TGF-β, Smad3 and the process of progressive fibrosis. Biochem Soc Trans 35(4): 661-664.
-
Zhu Y, Zhou J, Tao G (2011) Molecular aspects of chronic radiation enteritis. Clin Invest Med 34(3): E119-E124.
-
Gressner OA, Gressner AM (2008) Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int 28(8): 1065-1079.
-
Andrae J, Gallini R, Betsholtz C (2008) Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22(10): 1276-1312.
-
Dovedi SJ, Adlard AL, Bhalla GL, McKenna C, Jones S, et al. (2014) Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 74(19): 5458-5468.
-
Chen MF, Chen PT, Chen WC, Lu MS, Lin PY, et al. (2016) The role of PD-L1 in the radiation response and prognosis for esophageal squamous cell carcinoma related to IL-6 and T-cell immunosuppression. Oncotarget 7(7): 7913-7924.
-
Azad A, Lim SY, Costa ZD, Jones K, Diana A, et al. (2017) PD‐L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med 9(2): 167-180.
-
Ribas A (2015) Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov 5(9): 915-919.
-
Shin DS, Zaretsky JM, Ordinas HE, Diaz AG, Lieskovan SH (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7(2): 188-201.
-
Diaz AG, Shin DS, Moreno BH, Saco J, Ordinas HE, et al. (2017) Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 19(6): 1189-1201.
-
Box CV, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, et al. (2017) DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8(1): 15618.
-
Zhang N, Zeng Y, Du W, Zhu J, Shen D, et al. (2016) The EGFR pathway is involved in the regulation of PD- L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol 49(4): 1360-1368.
-
Gao SP, Mark KG, Leslie K, Pao W, Motoi N, et al. (2007) Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 117(12): 3846-3856.
-
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, et al. (2017) DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nature communications 8: 1751.
-
Permata TBM, Hagiwara Y, Sato H, Yasuhara T, Oike T, et al. (2019) Base excision repair regulates PD-L1 expression in cancer cells. Oncogene 38(23): 4452-4466.
-
Iijima M, Okonogi N, Nakajima NI, Morokoshi Y, Kanda H, et al. (2020) Significance of PD-L1 expression in carbon-ion radiotherapy for uterine cervical adeno/ adenosquamous carcinoma. J Gynecol Oncol 31(2): e19.
-
Sun LL, Yang RY, Li CW, Chen MK, Shao B, et al. (2018) Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res 8(7): 1307-1316.
-
Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, et al. (2018) ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J Clin Invest 128(9): 3926-3940.
-
Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, et al. (2021) Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell 39(10): 1388-1403.
-
Chiblak S, Tang Z, Lemke D, Knoll M, Dokic I, et al. (2019) Carbon irradiation overcomes glioma radioresistance by eradicating stem cells and forming an antiangiogenic and immunopermissive niche. JCI insight 4(2): e123837.
-
Yeo AT, Charest A (2017) Immune checkpoint blockade biology in mouse models of glioblastoma. J Cell Biochem 118(9): 2516-2527.
-
Yeung TL, Lazorchak A, Yu H, Qi J, Qin G, et al. (2021) Bintrafusp alfa enhances the antitumor efficacy of anti- VEGF and VEGF receptor kinase inhibitors in preclinical solid tumor models. Cancer Res 81(13): 443.
-
Dijke PT, Ichijo H, Franzen P, Schulz P, Saras J, et al. (1993) Activin receptor-like kinases: a novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene 8(10): 2879-2887.
-
Hsu C, Chang YF, Yen CJ, Lu LC, Zhu X, et al. (2021) Safety and efficacy of combination of GT90001, an anti- activin receptor-like kinase-1 (ALK-1) antibody, and nivolumab in patients with metastatic hepatocellular carcinoma (HCC). Journal of Clinical Oncology 39(3): 326.
-
Crocenzi TS, Khoueiry ABE, Yau T, Melero I, Sangro B, et al. (2017) Nivolumab in sorafenib-naive and- experienced patients with advanced hepatocellular carcinoma: CheckMate 040 study. J Clin Oncol 35(15): 4013.
-
Lazorchak A, Yu H, Qin G, Qi J, Marelli B, et al. (2021) Bintrafusp alfa enhances anti-tumor efficacy in combination with radiation therapy plus the ataxia telangiectasia mutated (ATM) inhibitor, M4076. Clinical Cancer Research 27(8): PO-088.
- Are the Vaccines the Only Solution to Prevent the COVID-19 Pandemic? Part Two
- Clinical Characteristics of Women in this New Global Immunodeficiency
- Cell Dynamics in HIV Pathogenesis: Insights and Implications
- Determination of the CDR (CDR1, CDR2) « Complementary- Determining Region Invertebrate Primitive Antibody from Sea Star »
- Prioritizing Care for High-Risk COVID-19 Patients in the EU: 10 Civic Recommendations to the Institutions
- Comprehensive Insights into ModRNA Vaccines: Persistent PP-Spike Recombinant Protein, Hyperimmune/Inflammatory Reactions, Thrombotic Vasculopathy, Chronic Organ Complications and Excess Deaths