Bioequivalence Study of Etoricoxib 120 mg in Healthy Subjects
Etoricoxib is an oral selective cyclo-oxygenase-2 (COX-2) inhibitor with anti-inflammatory and antirheumatic properties belonging to the group of NSAIDs. Study Objective: The objective of this study was to investigate the bioequivalence study of Etoricoxib, Tricox® 120 mg film coated caplet manufactured by PT Guardian Pharmatama for PT Nulab Pharmaceutical Indonesia in comparison with Etoricoxib 120 mg, Arcoxia® film coated tablet manufactured by Frosst Iberica, S.A., Spain, registered and packed by PT Merck Sharp Dohme Pharma Tbk Pasuruan, East Java. Indonesia. Methods: The study was conducted using an open-label, randomized, single-dose, two-periods, two-treatment, crossover study under fasting for 10 hours with 7 (seven) days washed-out period between each period. A single oral dose of the test drug or reference drug was administered to 16 healthy male subjects. The number of subjects who finished the study was fourteen (14) healthy male subjects. Serial plasma samples were obtained over a 72 hours period. Plasma concentrations of the drug were determined by LC-MS/MS method. From the Etoricoxib concentration vs. time curves, the following pharmacokinetic parameters were obtained: AUC0-72h, AUC0-∞, and Cmax, while the statistical interval proposed was 80.00 - 125.00% for AUC0-72h and Cmax with 90% Confidence Interval (CI) with α = 5.00%. The estimation of Tmax and T1/2 in the bioequivalence study was based on a nonparametric statistical procedure on the original data using Wilcoxon Sign Test. Results: The main pharmacokinetic parameters of the test drug Etoricoxib (BN: T200909) compared to reference drug, Arcoxia® (BN: T015857) were calculated based on geometric mean ratio and 90% confidence interval (CI). The results for AUC0-72h and Cmax were 91.97% (87.50% – 96.66%) and 96.98% (88.41% – 106.36%) respectively, with intra-subject variability (%CV) were 13.56% for AUC0-72h and 7.28% for Cmax. Hence, the number of 14 (fourteen) subjects has adequate number for required power of study. Conclusion: The study demonstrated that the test drug Etoricoxib (BN: T200909) manufactured by PT Guardian Pharmatama for PT Nulab Pharmaceutical Indonesia bioequivalence in term of both rate and extent of absorption to the reference drug Arcoxia® (BN: T015857) manufactured by Frosst Iberica, S.A., Spain, registered and packed by PT Merck Sharp Dohme Pharma Tbk Pasuruan, East Java. Indonesia.
Introduction
Etoricoxib is an oral selective cyclo-oxygenase-2 (COX- 2) inhibitor with anti-inflammatory and antirheumatic properties belonging to the group of Non-Steroid Anti- Inflammation Drugs (NSAIDs). It is indicated for symptomatic pain relief of osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, and acute gouty arthritis, and for short-term treatment of moderate pain associated with dental surgery [1].
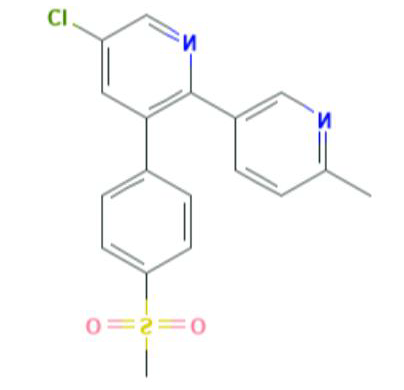
Etoricoxib (CAS 202409-33-4) is described chemically as 5-chloro-2-(6-methylpyridine-3-yl)-3-(4-methylsulfonyl phenyl) pyridine. The empirical formula is C18H15ClN2O2S. The molecular weight of Etoricoxib is 358.8 g/mol [2]. The solubility of Etoricoxib in water is 0.0033 g/L in water [3].
Etoricoxib is a member of a new class of agents called Coxibs. Etoricoxib is a potent, orally active cyclooxygenase-2 (COX-2) specific inhibitor within, and significantly above, the clinical dose range. Two isoforms of cyclooxygenase have been identified: cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). COX-1 is responsible for prostaglandin-mediated normal physiologic functions such as gastric cytoprotection and platelet aggregation. Inhibition of COX-1 by nonselective NSAIDs has been associated with gastric damage and inhibition of platelet aggregation. COX-2 is the isoform of the enzyme that has been shown to be induced by pro-inflammatory stimuli and has been postulated to be primarily responsible for the synthesis of prostanoid mediators of pain, inflammation, and fever. COX-2 is also involved in ovulation, implantation and closure of the ductus arteriosus, regulation of renal function, and central nervous system functions (fever induction, pain perception, and cognitive function). It may also play a role in ulcer healing. COX-2 has been shown to be primarily responsible for the synthesis of prostanoid mediators of pain, inflammation, and fever. Selective inhibition of COX-2 by etoricoxib (within the clinical dose range) decreases these clinical signs and symptoms with decreased potential for gastrointestinal (GI) toxicity and effects on platelet aggregation [4, 1].
Orally administered etoricoxib is well absorbed. The absolute bioavailability is approximately 100%. Etoricoxib is extensively metabolized with < 1 % of a dose recovered in urine as the parent drug. The major route of metabolism to form the 6’-hydroxymethyl derivative is catalyzed by CYP enzymes. CYP3A4 appears to contribute to the metabolism of etoricoxib in vivo. In vitro studies indicate that CYP2D6, CYP2C9, CYP1A2, and CYP2C19 also can catalyze the main metabolic pathway, but their quantitative roles in vivo have not been studied [5, 6].
Elimination of etoricoxib occurs almost exclusively through metabolism followed by renal excretion. Steady state concentrations of etoricoxib are reached within seven days of once daily administration of 120 mg, with an accumulation ratio of approximately 2, corresponding to a half-life of approximately 22 hours. The plasma clearance after a 25-mg intravenous dose is estimated to be approximately 50 ml/ min [5].
Etoricoxib is classified as a BCS II molecule. Etoricoxib exhibit very fast and complete dissolution in pH 2.0 media while dissolution, as expected, is slower at pH 4.5 and pH 6.8 [7]. There is a great need to develop technologies for these ‘BCS’ class II drugs for enhancing their dissolution rate, bioavailability and their formulation development. The enhancement of dissolution rate and oral bioavailability of poorly soluble drugs remains one of the most challenging aspects of drug product development [8].
The objective of this study is to investigate the bioequivalence of Etoricoxib 120 mg (Tricox®) manufactured by PT Guardian Pharmatama for PT Nulab Pharmaceutical Indonesia to the comparator drug (Arcoxia®) manufactured by Frosst Iberica, S.A., Spain, registered and packed by PT Merck Sharp Dohme Pharma Tbk Pasuruan, East Java. Indonesia.
Study Protocol
The study protocol was reviewed and approved by the Ethics Committee of the Medical Faculty University of Indonesia and the Indonesian Food and Drug Regulatory
Authority (BPOM RI).
The protocol described all details of the project, including design of the study, clinical procedures, bioanalysis of blood samples obtained from the participants, pharmacokinetic and statistical data analysis, bioequivalence evaluation, the informed consent form, and ultimately documentation and final report issuance.
Ethical Considerations
The study was carried out according to The International Council for Harmonization (ICH) guidelines for Good Clinical Practice (GCP) and the declaration of Helsinki provisions [9, 10]. The Institutional Review Board (IRB) which issued ethical approval for this study was the Ethics Committee of the Medical Faculty University of Indonesia which was certified by the Forum for Ethical Review Committee in the Asia and Western Pacific Region (FERCAP). Each subject was given the informed consent form during the screening phase prior to the study. A meeting was arranged by the principal investigator and clinical investigators to explain all details of the study to the subject, including the purposes, risks, advantages, procedures, and the right as a research subject to withdraw at any time during the study, and the compensation in case of any harm caused by the study.
Study Design
This study was conducted in an open-label, randomized, single-dose, two-periods, two-treatment, and crossover study under fasting conditions [4]. The subjects were randomly assigned to each dosing sequence of the investigational drug products (test and reference formulations). This study was carried out on sixteen (16) healthy subjects, where subjects S12 and S14 dropped out of the study in the 2nd period because of personal reasons.
Inclusion and Exclusion Criteria for Participation in the Study
The subjects were regarded as eligible for participation in this study based on the following inclusion criteria: willing to sign an informed consent, adult male/female subjects with ages between 18-55 years, and body mass index (BMI) ranging from 18–25 kg/m2, vital sign after 10 minutes resting within ranges: pulse rate 60-90 bpm, respiratory rates 12- 20 x/minutes, systolic blood pressure 100-130 mmHg and diastolic blood pressure 60-90 mmHg, the subject must have 12 lead electrocardiogram (ECG) without any significant abnormalities, and negative result of rapid test antigen Covid 19 in 1st and 2nd period.
The exclusions criteria including participate in another study within 3 months prior to the first day of study drug administration, pregnant or lactating female, smokers and smoking more than 10 cigarettes per day, intake of any prescription drug or non-prescription drug within 7 days prior the first day of drug administration of this study, blood donation or blood loss of 300 mL (or more) within 3 months prior to the first day of study drug administration, history of drug and/or alcohol abuse or dependency within 12 months prior to the first day of study drug administration, known hypersensitivity or contraindication to the study drug, any surgical or medical condition (present or history) which might significantly alter the absorption, distribution, metabolism or excretion of the study, e.g. gastrointestinal disease including gastric or duodenal ulcers or history of gastric surgery, history of any bleeding or coagulative disorders, clinically significant hematology abnormalities, clinically significant urinalysis abnormalities, renal insufficiency (plasma’s creatinine concentration ≥ 1.50 mg/ dL), history or presence of any liver dysfunction (SGPT, alkaline phosphate, total bilirubin ≥ 1.5 ULN), and positive result of HBsAg, HCV, and/ or HIV test.
Health Screening
Health screening is conducted prior to the study to evaluate the subject’s health condition based on inclusion and exclusion criteria. Subjects were through medical examination within 7 days prior to their first study drug administration day. These include assessment of physical examination, vital signs (i.e. blood pressure, pulse rate, and body temperature), and ECG was conducted by Responsible Physician in Biometrik Riset Indonesia. Laboratory values of liver function (AP, SGPT, SGOT, and total/direct bilirubin), renal function (serum creatinine and urea), routine hematology (hemoglobin, hematocrit, erythrocytes, platelets leucocyte count, leukocyte differential count, and erythrocytes sedimentation rate), blood glucose, routine urinalysis (specific gravity, pH, leucocytes esterase, nitrite, albumin, glucose, ketones, urobilinogen, bilirubin, occult blood, tubular and sediment) were tested by Clinical Laboratory. Immunology test for HBsAg, HCV, HIV, and Rapid Test Antigen Covid-19 was conducted in Biometrik Riset Indonesia. During the screening and immunology test (HBsAg, HCV, and HIV) approximately 10 mL of blood samples were drawn from each subject
Drug Product Administration
The subjects were admitted to the Biometrik Riset Indonesia on a day before the study for quarantine. All subjects were fasted for 10 hours overnight starting from 9 p.m. to 7 a.m. to drug administration. At 7 p.m. on the sampling day, subjects were instructed to consume one film- coated caplet of the test drug or one film-coated tablet of the reference drug.
A single oral dose of the study drug was given in a pill case, swallowed by each subject with 250 mL of water according to randomization. After 2 hours, warm water was provided as desired. No food was allowed until 4 hours after study drug administration. Standard meals were reserved for 4 hours (breakfast), 6 hours (lunch), and 12 hours (dinner) after study drug administration. Subjects were remaining in a sitting position until 4 hours period after drug administration. Subjects were not allowed to exit the clinical facility except with the Responsible Physician's permission.
**Blood Samples Collection**
After fasting overnight, approximately at 6 a.m. on the sampling day, a 5 mL pre-dose blood sample was taken within one hour prior to drug administration. After drug administration, 5 mL blood samples were taken at 0.17, 0.33, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, and 72 hours drug administration. After a washout period of seven (7) days, subjects underwent the second period of the study. The procedure of the second period was repeated in the same manner to complete the crossover design.
Blood samples were drawn with a disposable syringe and transferred into K3EDTA blood collection tubes. Plasma was separated by centrifugation at 7,000 rpm (≈ 6,793 x g) for 5 minutes and immediately transferred plasma into three clean microtubes 2.0 mL which consist of two microtubes for analysis and one microtube for retained sample. Plasma separation was carried out in a plasma separation room with a temperature range of 20 - 30°C, and a humidity range of 40 - 70%.
Approximately the total volume of blood samples collected from each subject is 190 mL during screening and two periods of sampling, which is consisting of 10 mL of blood samples taken for screening and 180 mL of blood samples that were taken from each subject during two periods of sampling.
**Statistical Analysis**
The statistical method for testing bioequivalence is ANOVA for 2-treatment, 2-sequence, 2-period, cross-over comparing AUC${0-72h}$ and $C{\text{max}}$ after transformation of the original into their logarithmic values where the factors formulation, period, sequence, and subject nested within the sequence was used to explain overall variability in the observations. The acceptance criterion of the bioequivalence study is the value of 90% confidence interval with $\alpha = 5.00\%$ of the Test/Reference geometric means ratio must be in the range of 80.00-125.00% for AUC${0-72h}$ and $C{\text{max}}$ [11, 12, 13].
EquivTestPK software was used to perform the statistical analysis of AUC${0-72h}$, AUC${0-90}$, and $C_{\text{max}}$ using analysis of variance (ANOVA) after transformation of the data to their logarithmic (In) values. Using the error variance (S2) obtained from the ANOVA; the 90% CI with $\alpha = 5.00\%$ were calculated from the following equation:
$$90\% \text{CI} = \left( \bar{X}_T - \bar{X}_R \right) \pm t_{0.1}(\nu) \sqrt{S^2 \frac{1}{n_{\text{TR}}} + \frac{1}{n_{\text{TR}}}}$$
- $\bar{X}_T, \bar{X}_R$: the means of in transformed values for the test drug [T] and the reference drug [R].
- S2: the error variance obtained from the ANOVA.
- nTR, nRT: the number of subjects of sequence TR and RT.
- t0.1: the t-value for 90% CI with $\alpha = 5.00\%$.
- v: the degree of freedom of the error variance from ANOVA.
The anti (In) of the above confidence intervals (CIs) are the 90% CIs with $\alpha = 5.00\%$ of the ratios of the test/the reference geometric means. The difference in $T_{\text{max}}$ was analyzed non-parametrically on the original data using Wilcoxon Test. Meanwhile, $T_{1/2}$ was analyzed using parametric or non-parametric statistics on the original data (not transformed) depending on whether the data is normally distributed or not using Wilcoxon Test [13].
**Assay Methodology and Validation**
Prior to the assay of Etoricoxib in the sample, bioanalytical method validation was evaluated for anticoagulant effect (i.e. comparing the effect of CPDA anticoagulant used in blank plasma bought from Indonesian Red Cross for method validation towards anticoagulant used in K3EDTA anticoagulant used in blood collection tube to collect blood samples); selectivity; carry-over effect; calibration curve and Lower Limit of Quantification (LLOQ); precision and accuracy; matrix effect; dilution integrity; and stabilities (i.e. short term stability at room temperature and post-preparative/autosampler batch integrity, freeze-thawed stability, also long term stability). An assay of Etoricoxib concentration in plasma was carried out by a fully validated LC MS/MS with LLOQ 10.00 ng/mL.
During the bioanalytical phase of the plasma samples, the analysis was monitored by the quality control process include system suitability test, linearity of calibration curve, and quality control samples (Low QC, Medium QC, and High QC) referred to requirement described in European Medicinal Agency (EMA) guideline 2011[11].
Data Quality Assurance
Drug accountability is maintained by investigators who keep all the records of the disposition of all study drugs received, administered, accidentally destroyed, and destroyed when expired. At the end of the study, all unused study drugs will be kept in the study site and will be destroyed after one (1) years from the final reports received by the Sponsor, six (6) months after the study drug shelf-life (study drug’s expired date) or registration permit number (NIE) accommodates the request of re- review by Indonesian Food & Drug Regulatory Authority (BPOM RI) [13]. The rest of the plasma sample analysis was destroyed following stability long-term validated method, while retain sample will be destroyed for at least 1 year after the final reports had been sent to the Sponsor. The data handling and verification were maintained by making Case Report Form (CRF) which was filled legibly using a blue ballpoint. The forms were verified against all original records. A copy was retained in the investigator’s files, and all other copies were given to the Sponsor. Sponsor generally performs site/clinical monitoring of clinical trials to assure high-quality trial conduct. Therefore Sponsor was appointing their personnel as the Monitor of the study. The Monitor is committed to professional secrecy. They perform monitoring of individual case histories, assess adherence to the study protocol, ensure the ongoing implementation of appropriate data entry and quality control procedures, and in general assess adherence to Good Clinical Practices [14].
Results and Discussion
The total number of subjects who finished the study was 14 subjects, where subject S12 and S14 dropped out of the study in the 2nd period because of personal reasons. The demographic data of the subject are tabulated in Table 1.
| MIN | MAX | |
|---|---|---|
| Age (year) | 18 | 48 |
| BMI (kg/m2) | 18.2 | 25 |
| Pulse (bpm) | 62 | 90 |
| Respiratory Rate (x/minute) | 14 | 18 |
| Blood Pressure (mm/Hg) | 100/70 | 130/90 |
Table 2: Demographic Data of 14 subjects.
There is no adverse event occurred during this study.
Bioanalytical Result
Results of applying the bioanalytical method to the bioequivalence study of Tricox® (BN: T200909) manufactured by PT Guardian Pharmatama for PT Nulab Pharmaceutical Indonesia in Fourteen (14) healthy male subjects, were all calibration curves of the subject showed good linearity within the range of ng/mL with the coefficient of correlation (R2) ≥ 0.9929. For Accuracy and precision provided on Table 2.
| Accuracy | Precision | |
|---|---|---|
| QC Low (30.06 ng/mL) | 0.79% | 5.49% |
| QC Med (2,505.01 ng/mL) | 3.84% | 2.84% |
| QC High (3,757.52 ng/mL) | 2.84% | 5.97% |
| LLOQ (9.98 ng/mL) | 1.28% | 1.38% |
Table 1: Accuracy and Precision.
Pharmacokinetic Analysis
The pharmacokinetic parameters (AUC0-72h, AUC0-∞, Cmax) of test drug (T) and reference drug (R) were calculated and compared to assessed bioequivalence. The calculated 90% CI with α = 5.00% for geometric mean of individual and the ratios of AUC0-72h and AUC0-∞ as well as Cmax for the test drug: Tricox® (BN: T200909), and reference drug: Arcoxia® (BN: T015857) were all within 80.00 - 125.00% interval. This was in conformity with the standard guideline for bioequivalence study [13].
The result of Tmax and T1/2 shown in Table 3. Meanwhile, the main pharmacokinetic parameters drug of study Etoricoxib was obtained from 14 subjects after oral administration of test drug and reference drug shown in Table 4.
The means of plasma concentrations vs. time profiles after a single dose of oral administration of investigational products are shown in Figure 2.
| Parameter | Test | Reference |
|---|---|---|
| T (hours) max | 1.50 (0.75 – 4.00) | 1.75 (1.00 - 4.00) |
| T (hours) 1/2 | 18.80 ± 7.65 | 19.20 ± 8.00 |
Table 3: The Result of Tmax and T1/2.
![Figure 2: Geometric Means of Plasma Concentration vs. Time Profiles after Dosing of Test Drug [T]: Tricox® and Reference Drug [R]: Arcoxia®.](/fulltextimages/8926/fig_2.png)
| Parameter | Mean (SD) | Geometric Mean Ratio of T/R (90% CI) | % Intrasubject CV | Statistical Test Reference Power (%) | |
|---|---|---|---|---|---|
| Test | Reference | ||||
| AUC0-72h (ng.h/mL) | 40.685,40 (10.219,72) | 44.950,17 (12.056,94) | 91.97% (87.50 – 96.66)% | 7.28% | 100% |
| AUC0-∞ (ng.h/mL) | 45.186,58 (13.670,35) | 49.983,32 (15.245,06) | 91.48 % (86.50 - 96.74)% | 8.19%. | 100% |
| Cmax (ng/mL) | 2.472,79 (384,24) | 2.536,95 (419,38) | 96.98% (88.41 – 106.36)% | 13.56% | 100% |
Table 4: Pharmacokinetic Parameters of Etoricoxib after a Single-Dose Oral Administration of Test & Reference Drug.
Conclusion
Based on the data obtained from this study, it is concluded that the test drug Tricox® (BN: T200909) manufactured by PT Guardian Pharmatama for PT Nulab Pharmaceutical Indonesia BIOEQUIVALENCE in terms of both rate and extent of absorption to the reference drug Arcoxia® (BN: T015857) manufactured by Frosst Iberica, S.A., Spain, registered and packed by PT Merck Sharp Dohme Pharma Tbk Pasuruan, East Java, Indonesia with geometric mean ratio and 90% confidence interval for AUC0-72h and Cmax parameter were 91.97% (87.50%-96.66%) and 96.98% (88.41%-106.36%) respectively (requirement 80.00 - 125.00% for AUC0-t and Cmax) and the intra-subject coefficient of variation is 7.28% and 13.56%. Therefore, it can be concluded that the two formulations are therapeutically equivalent and interchangeable.
References
-
(2020) Compound Summary: Etoricoxib. PubChem
-
(2019) Decentralised Procedure: Public Assessment Report Etoricoxib Zydus
-
(2020) Etoricoxib Human Metabolome Data Base: Showing Metabocard for Etoricoxib (HMDB0015565).
-
Shohag MH, Islam MS, Ahmed MU, Joti JJ, Islam MS, et al. (2011) Pharmacokinetic and Bioequivalence Study of Etoricoxib Tablet in Healthy Bangladeshi volunteers. Arzneimittelforschung 61(11): 617-621.
-
(2018) Guidance Document: Conduct and Analysis of Comparative Bioavailability Study. Health, Canada.
-
(2019) Arcoxia: Australian Product Information. MK0663-AUS-2017-015150, Merck Sharp & Dohme Pharma Corporation.
-
Mitra A, Kesisoglou F, Dogterom P (2015) Application of Absorption Modeling to Predict Bioequivalence Outcome of Two Batches of Etoricoxib Tablets. AAPS PharmSciTech 16(1): 76-84.
-
Chowdary KPR, Reddy MV (2011) Formulation Development Studies on Enhancement of Solubility and Dissolution Rate of Etoricoxib by Cyclodextrin Complexation. Asian Journal of Chemistry 23(4): 1445- 1448.
-
(2015) Guidelines for Good Clinical Practices. Indonesian Food & Drug Regulatory Authority (BPOM RI).
-
(2008) The Declaration of Helsinki. Bulletin of the World Health Organization. 86: 652.
-
(2011) Guideline on Bioanalytical Method Validation. European Medicines Agency.
-
(2010) Guideline on the Investigation of Bioequivalence. European Medicines Agency.
-
(2015) Bioequivalence Study Guidelines. Indonesian Food & Drug Regulatory Authority (BPOM RI).
-
(2006) Guidance for Clinical Trial Sponsors - Establishment and Operation of Clinical Trial Data Monitoring Committees. US Department of Health and Human Services, Food and Drug Administration, USA.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review