Bioequivalence Study Comparing a Generic and Innovator Drug of Rosuvastatin 20 mg
The bioequivalence study was conducted to compare the bioavailability of two rosuvastatin 20 mg film-coated tablet formulations (test and reference formulation). This study was an open-label, randomized, single-dose, two-periods, twotreatments, and crossover study which included 32 healthy adult male and female subjects under fasting conditions. Each of the two study periods was separated by a 7 days washout period. A single oral dose of test or reference drug was administered to the subject in each period based on the randomization scheme. Plasma concentrations of the drug were determined by LC-MS/MS method. The pharmacokinetic parameters assessed in this study were the area under the plasma concentration-time curve from time zero to 72 h (AUC0-72h), area under the plasma concentration-time curve from time zero to infinity (AUC0-∞), the peak plasma concentration of the drug (Cmax), time needed to achieve the peak plasma concentration (Tmax), and the elimination half-life (T1/2). The geometric mean ratio (GMR) and 90% Confidence Interval (90% CI) for AUC0-72h and Cmax of test/reference drug for rosuvastatin were 97.05 % (89.07%– 105.74%) and 101.15% (89.53%– 114.26%). Since the 90% CI with α 0.05% for AUC0-72h and Cmax of rosuvastatin were within the standard bioequivalence range (80.00– 125.00%), it was concluded that the two rosuvastatin film-coated tablets (test and reference drug) were bioequivalence in terms of the rate and extent of absorption.
Introduction
Bioequivalence (BE) studies is a study that evaluates the equivalence between a generic drug and an innovator drug [1]. Therefore, in this study, we compared two formulations of rosuvastatin film-coated tablets i.e. the innovator drug or which we called the reference drug, and a generic drug which we called the test drug. Rosuvastatin is indicated for the treatment of adult patients with hypertriglyceridemia, as indicated as adjunctive therapy to diet to reduce elevated Total-C, LDL-C, ApoB, nonHDL-C, and triglycerides and to increase HDL-C in adult patients with primary hyperlipidemia or mixed dyslipidemia [2].
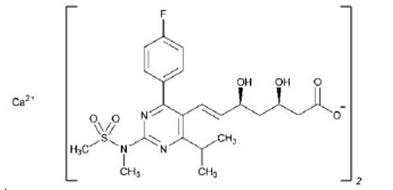
Rosuvastatin is also known as a synthetic HMG-CoA reductase inhibitor. More specifically, it is produced as Calcium (3R,5S,E)-7-(4-(4-fluorophenyl)-6-isopropyl- 2-(N-methylmethylsulfonamido)pyrimidin-5-yl)- 3,5-dihydroxyhept-6-enoate salt) with the empirical formula Ca(C22H27FN3O6S)2 [3]. Rosuvastatin has a few characteristics such as white to off-white powder, being freely soluble in acetonitrile, sparingly soluble in water and methanol, and slightly soluble in ethanol, and has a molecular weight of 1001.14 g/mol [4, 5].
In clinical pharmacology studies, peak plasma concentrations of rosuvastatin were reached 3 to 5 hour following oral dosing. Both Cmax and AUC increased in approximate proportion to the rosuvastatin dose. The absolute bioavailability of rosuvastatin is approximately 20%. Administration of rosuvastatin with food did not affect the AUC of rosuvastatin. The AUC of rosuvastatin does not differ following evening or morning drug administration [2].
Rosuvastatin is taken up extensively by the liver which is the primary site of cholesterol synthesis and LDL-C clearance. The mean volume of distribution at the steady state of rosuvastatin is approximately 134 liters. Rosuvastatin is 88% bound to plasma proteins, mostly albumin. This binding is reversible and independent of plasma concentrations. Rosuvastatin is not extensively metabolized; approximately 10% of a radiolabeled dose is recovered as a metabolite. The major metabolite is N- desmethyl rosuvastatin, which is formed principally by cytochrome P450\2C9, and in vitro studies have demonstrated that N-desmethyl rosuvastatin has approximately one-sixth to one-half the HMG-CoA reductase inhibitory activity of the parent compound. Overall, greater than 90% of active plasma HMG-CoA reductase inhibitory activity is accounted for by the parent compound [2].
Following oral administration, rosuvastatin and its metabolites are primarily excreted in the feces (90%). Approximately 5% is excreted unchanged in the urine. After an intravenous dose, approximately 28% of total body clearance was via the renal route, and 72% by the hepatic route. Rosuvastatin is primarily eliminated by excretion in the feces. The elimination half-life of rosuvastatin is approximately 19 hours. As with other HMG-CoA reductase inhibitors, the hepatic uptake of rosuvastatin involves the membrane-transported OATP-C. This transporter is important in the hepatic elimination of rosuvastatin [2].
Subjects, Materials and Methods
Subjects and Study Design
Thirty-two (32) healthy subjects, which consist of twenty- six (26) male and six (6) female subjects, 20-48 years old, and Body Mass Index (BMI) between 18.42 kg/m2 – 24.88 kg/m2 were enrolled in the study. All subjects pass the screening and selection phase with inclusion and exclusion criteria based on Covid- 19 testing, physical examination, vital signs (blood pressure, pulse/heart rate, respiratory rate, and body temperature), electrocardiogram (ECG), blood biochemistry including glucose, liver function (AP, SGPT, SGOT, and total/ direct bilirubin), renal function (serum creatinine, and urea), sero-immunology (HBsAg, anti- HCV, and anti-HIV), routine hematology (hemoglobin and leucocyte), blood glucose, and urinalysis (specific gravity, pH, leukocyte esterase, nitrite, albumin, glucose, ketones, urobilinogen, bilirubin, occult blood, tubular and sediment). A pregnancy test was performed for female subjects in the screening phase and before drug administration in each period.
This study was an open-label, randomized, single-dose, two-periods, two-treatments, cross-over study in fasting conditions with 7 days of washout between each period. The study was performed in accordance with The International Council for Harmonization (ICH) guidelines for Good Clinical Practice (GCP) and the declaration of Helsinki provisions [6, 7]. This study was also approved by the Indonesian Food and Drug Regulatory Authority and the Ethics Committee of the Medical Faculty University of Indonesia which was certified by the Forum for Ethical Review Committee in the Asia and Western Pacific Region (FERCAP).
Treatment Phase and Blood Sampling
Subjects attended PT Biometric Riset Indonesia a night before drug administration and conducted rapid test antigen for Covid-19 in each period, only the subject with a negative result of Covid-19 was allowed to proceed in this study. The subjects were fasting overnight from 9 p.m. until 7 a.m. A 5 mL blood sample (pre-dose) was taken one hour before drug administration for each period. One film-coated tablet of the study drug, whether its test or reference drug was given at 7 a.m. with 240 mL of water.
After drug administration, 5 mL of blood was taken at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 9, 12, 16, 24, 36, 48, and 72 hours for each period. Seven days after the first drug administration (washout period), the same procedure was repeated with the alternate drug to complete the crossover design. Plasma was separated from the blood samples by centrifuging at 4000 rpm for 10 minutes in a room with a temperature range of 20 - 24°C and humidity range of 65 - 68%. Samples for analysis were stored at (-20) oC and the retained samples were stored at (-80)°C.
The date and the time of drug administration (dosing) and taking of each blood sample (sampling) were recorded in the Case Report Form (CRF) for each subject.
Statistical Analysis
EquivTestPK software was used to perform the statistical analysis of AUC0-72h, AUC0-∞, and Cmax using analysis of variance (ANOVA) after the transformation of the data to their logarithmic (ln) values. The anti (ln) confidence intervals (CIs) are the 90% CIs with α = 5.00% of the ratios of the test/ the reference geometric means. The power of the study should be 80% with α = 5.00%. The acceptance criterion of the bioequivalence study is the value of a 90% confidence interval with α = 5.00% of the test/reference geometric means ratio must be in the range of 80.00-125.00% for AUC0-
72h, and Cmax [8, 9, 10]. The differences between drugs (T/R) in Tmax and T1/2 parameters were analysed nonparametrically on the original data using Wilcoxon Test.
Assay Methodology and Validation
Prior to the assay of rosuvastatin in the sample, bioanalytical method validation was carried out by a fully validated LC-MS/MS and evaluated for anticoagulant effect (i.e. comparing the effect of CPDA anticoagulant used in blank plasma bought from Indonesian Red Cross for method validation towards anticoagulant used in K3EDTA anticoagulant used in blood collection tube to collect blood samples); selectivity; carry-over effect; calibration curve and Lower Limit of Quantification (LLOQ); precision and accuracy; matrix effect; dilution integrity; and stabilities (i.e. short term stability at room temperature and post- preparative/autosampler batch integrity, freeze-thawed stability, also long term stability). Bioanalytical method validation was conducted and meets the requirements of the European Medicinal Agency (EMA) Validation Guideline 2011.
Sample Preparation and Analysis
An amount of plasma sample was added with internal standard Etoricoxib. The sample was precipitated using ethyl acetate. The organic phase was injected into a liquid chromatography system. The analytical column was Agilent Poroshell 120 EC-C-18 (2.7 µm) 3.0 mm x 50 mm. The mobile phase used was a mixture of acetonitrile: formic acid 0.20% (60:40 v/v) and the injection volume was 10 µL, with a flow rate of 0.20 mL/min.
An assay of rosuvastatin concentration in plasma was carried out by LC-MS/MS with LLOQ 0.10 ng/mL. During the bioanalytical phase of the plasma samples, the analysis was monitored by the quality control process including system suitability test, linearity of a calibration curve in the range 0.1 ng/mL - 100 ng/mL, and quality control samples (Low QC of 0.3 ng/mL, Medium QC of 50 ng/mL, and High QC of 75 ng/mL) referred to requirements described in European Medicinal Agency (EMA) guideline 2011 [11].
Result
There were 32 subjects (consisting of 26 male and 6 female subjects). The demographic data of the subjects enrolled in this study are tabulated in Table 1.
| Min | Max | |
|---|---|---|
| Age (year) | 20 | 48 |
| BMI (kg/m2) | 18.42 | 24.88 |
| Pulse (bpm) | 65 | 89 |
| Respiratory Rate (x/minute) | 16 | 16 |
| Blood Pressure (mm/Hg) | 100/62 | 129/84 |
Table 1: Demographic Data of the Subject.
The blood samples from 32 subjects were analyzed for plasma concentrations of rosuvastatin. Mean plasma concentrations versus time profiles of rosuvastatin in human subjects (n = 32) after oral administration of 20 mg rosuvastatin film-coated tablet of test drug and reference drug are shown in Figure 2.
![Figure 2: Geometric Means of Plasma Concentration vs. Time Profiles after Dosing of Test Drug [T]: Rosuvastatin and Reference Drug [R]: Crestor® The pharmacokinetic parameters (AUC0-72h, AUC0-∞, Cmax) of the test drug (T) and reference drug (R) were calculated and compared to assess bioequivalence. The calculated 90% CI with α = 5.00% for the geometric mean of individual and the ratios of AUC0-72h and AUC0-∞ as well as Cmax for the test drug: rosuvastatin (BN: 22H6015) and reference drug: Crestor® (BN: 60042201) were all within 80.00 – 125.00% interval.](/fulltextimages/10034/fig_2.png)
Figure 2: Geometric Means of Plasma Concentration vs. Time Profiles after Dosing of Test Drug [T]: Rosuvastatin and Reference Drug [R]: Crestor® The pharmacokinetic parameters (AUC0-72h, AUC0-∞, Cmax) of the test drug (T) and reference drug (R) were calculated and compared to assess bioequivalence. The calculated 90% CI with α = 5.00% for the geometric mean of individual and the ratios of AUC0-72h and AUC0-∞ as well as Cmax for the test drug: rosuvastatin (BN: 22H6015) and reference drug: Crestor® (BN: 60042201) were all within 80.00 – 125.00% interval.
This was in accordance with the standard guideline for bioequivalence studies [8, 9, 10]. The main pharmacokinetic parameters drug of study rosuvastatin was obtained from 32 subjects after oral administration of the test drug and the reference drug is shown in Table 2. Meanwhile, the result of Tmax and T1/2 is shown in Table 3.
| Mean (SD) | Geometric Mean Ratio of T/R (90% CI) (%) | % Intra- subject CV (%) | % Inter-subject CV (%) | |||
|---|---|---|---|---|---|---|
| Parameter | Test | Reference | Test | Reference | ||
| AUC (ng.h/mL) 0-72h | 273.44 (134.05) | 280.83 (146.75) | 97.05 (89.07 – 105.74) | 20.20 | 49.02 | 52.25 |
| C (ng/mL) max | 37.82 (21.38) | 36.20 (19.20) | 101.15 (89.53 – 114.26) | 28.74 | 56.52 | 53.02 |
| AUC (ng.h/mL) 0-∞ | 275.67 (134.60) | 283.24 (147.03) | 96.91 (88.96 – 105.56) | 20.15 | 48.83 | 51.91 |
Table 2: Pharmacokinetic Parameters of Rosuvastatin after a Single-Dose Oral Administration of Test & Reference Drug.
| Parameter | Test | Reference |
|---|---|---|
| T (hours) max | 2.50 (0.50 - 5.00) | 2.50 (1.50 - 5.00) |
| T (hours) 1/2 | 7.70 ± 1.57 | 7.69 ± 1.45 |
Table 3: The Result of Tmax and T1/2.
Discussion
A reliable bioequivalence study should be performed in order to ensure that a generic/test drug possesses similar quality as its reference, in terms of safety and efficacy. Therefore, a generic/test should be produced at more economical costs, but without compromising the quality.
Providing generic medicinal products supports pharmaco- economics, which benefits both patients and healthcare professionals since they have options to select quality medications at affordable prices [12, 13].
The aim of the study was to evaluate the bioequivalence of the test rosuvastatin film-coated tablet (BN: 22H6015)
and the reference rosuvastatin film-coated (Crestor® BN: 60042201) administered as 20 mg single oral dose. In order to eliminate the effect that food has on drug absorption, the formulations were administered to volunteers after overnight fasting.
Main parameters obtained in this study were AUC0-
72, Cmax, and AUC0-∞. The geometric mean ratio and 90% confidence interval for AUC0-72, Cmax, and AUC0-∞ parameters were 97.05% (89.07%– 105.74%), 101.15% (89.53%– 114.26%) and 96.91% (88.96% –105.56%). Confidence Intervals of the test/reference ratios for AUC0-72, AUC0-∞, and Cmax of rosuvastatin were within the acceptable range for bioequivalence (80 – 125 %).
The mean ratio of AUC0-72 /AUC0-∞ of rosuvastatin for both the test drug and the reference drug was greater than 80 % (99.19 % for the test drug and 99.15 % for the reference drug). This showed that the sampling time was long enough to guarantee an adequate description of the absorption phase.
The mean (SD) elimination half-life T½ of rosuvastatin for the test drug was 7.70 ± 1.57 h and for the reference drug it was 7.69 ± 1.45 h. Utilizing the Wilcoxon Signed Rank Test, the half-life values of the test and the reference drug were not significantly different, demonstrating a comparable rate of drug elimination from the body. The median (range) of the time to reach the maximum rosuvastatin plasma concentration (Tmax) of the test drug was 2.50 (0.50 - 5.00) h and 2.50 (1.50 - 5.00) h for the reference drug. Utilizing Wilcoxon Signed Rank Test, the Tmax of the two evaluated preparations did not different significantly.
In the present study, the intrasubject coefficient of variance (%CV) obtained from the ANOVA for the rosuvastatin AUC0-72 was 20.20% and Cmax 28.74%, meaning that the required sample size for at least 80 % power should be 32 subjects. It can be concluded that the power study was adequate for this study.
There was no dropout case during the study. Thirty- two (32) healthy subjects completed the study without adverse events encountered during this study.
After all study phases are completed, remaining samples and data management should be maintained. A copy of CRF and all source data were retained in the investigator’s files. Other copies were given to the Sponsor as needed. Additional consideration was made about complying with applicable local laws, guidelines, etc. Study document binders were provided for all required study documents.
Conclusion
Based on the statistical calculation of pharmacokinetic parameter results of this study, it is concluded that the test drug rosuvastatin (BN: 22H6015) manufactured by PT Otto Pharmaceutical Industries is bioequivalent in terms of both rate and extent of absorption with the reference drug Crestor® (BN: 60042201) manufactured by IPR Pharmaceuticals Inc. Puerto Rico for AstraZeneca UK Limited, United Kingdom; packaged and released by AstraZeneca Pharmaceutical Co. Ltd, China; imported by PT AstraZeneca Indonesia. Therefore, it can be concluded that the two formulations of rosuvastatin 20 mg are pharmacokinetically equivalent and interchangeable.
References
-
Lou Y, Jones MP, Sun W (2019) Estimation of causal effects in clinical endpoint bioequivalence studies in the presence of intercurrent events: noncompliance and missing data. Journal of biopharmaceutical statistics 29(1): 151-173.
-
AstraZeneca (2018) Highlight of Prescribing Information: Crestor® (rosuvastatin calcium) tablets.
-
Harahap Y, Prasaja B, Azmi F, Lusthom W, Sinandang T, et al. (2016) Bioequivalence study of two rosuvastatin tablet formulations in healthy Indonesian subjects. Int J Clin Pharmacol Ther 54(3): 212-216.
-
(2021). Certificate Rosuvastatin Calcium. The United States Pharmacopeial (USP) Convention. CERT1-07.
-
(2016) Public Assessment Report Rosuvastatin, UK.
-
(2015) Guidelines for Good Clinical Practices. Indonesian Food & Drug Regulatory Authority (BPOM RI).
-
Williams JR (2008) The Declaration of Helsinki and Public Health. Bull World Health Organ 86(8): 650-652.
-
(2022) Bioequivalence Study Guidelines. Indonesian Food & Drug Regulatory Authority (BPOM RI).
-
(2015) ASEAN Guidelines for the Conduct of Bioequivalence Studies pp: 1-27.
-
(2010) Guideline on the Investigation of Bioequivalence. European Medicines Agency.
-
(2011) Guideline on Bioanalytical Method Validation. European Medicines Agency.
-
Arenas-Guzman R, Tosti A, Hay R, Haneke E (2005) National Institue for Clinical Excellence. Pharmacoeconomics – an aid to better decision-making. J Eur Acad Dermatol Venereol 19(Suppl 1): 34-39.
-
Trask LS (2002) Pharmacoeconomics: principles, methods, and applications. In: DiPiro JT, et al. (Eds.), Pharmacotherapy: A Pathophysiologic Approach. 8th(Edn.), McGraw-Hill Education, LLC, New York, pp: 1-2.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review