A Unique Case of Primary Hepatic CD30-Positive, CD15-Negative Classical Hodgkin’s Lymphoma Presenting as Fever of Unknown Origin and Acute Hepatic Failure
Hodgkin’s lymphoma is a B-cell lymphoproliferative neoplasm that commonly presents with lymphadenopathy and constitutional symptoms. Primary Hodgkin’s lymphoma of the liver without extrahepatic lymphadenopathy is rare, representing about 0.016% of all non-Hodgkin’s lymphoma and 0.4% of all extranodal lymphomas. Most primary hepatic lymphomas (PHL) are of the diffuse large B-cell histology. In a patient presenting with fever and acute hepatic failure in the absence of extrahepatic lymphadenopathy, the diagnosis of primary Hodgkin’s lymphoma should be entertained. To our knowledge, this is the first reported case of CD30-positive, CD15-negative primary hepatic classical Hodgkin’s lymphoma.
Introduction
Hodgkin’s lymphoma (HL) is a B-cell lymphoproliferative neoplasm with a peak in diagnosis in young adulthood (ages 20-34). The estimated new cases of HL in 2017 is 8,260, and estimated deaths in 2017 from the disease is 1,070 [1]. It is a rare malignancy in the United States, accounting for 0.5% of all newly diagnosed malignancies.
Staging for HL is based on the Ann Arbor staging system [2], which involves the location, size and extent of lymphadenopathy and other lymphoid tissue, involvement of extra-lymphatic organs, and constitutional symptoms. Essential steps for obtaining an accurate stage in HL includes the following: obtaining a detailed history and physical examination, complete blood cell count, erythrocyte sedimentation rate (ESR), comprehensive metabolic panel including liver function tests, contrast- enhanced diagnostic computed tomography (CT), and positron emission tomography/computed tomography (PET/CT).
HL is categorized into two main groups: classical HL (comprising of four histologic subtypes: nodular sclerosing, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted) and nodular lymphocyte- predominant Hodgkin’s lymphoma (NLPHL). The classic neoplastic abnormal cell found on pathology in classical HL is the Reed-Sternberg (RS) cell, which is a large, mono- or multi-nucleated neoplastic B-cell derived from the lymph node germinal center admixed in a background of normal inflammatory cells. Classical HL cells immunophenotypically are positive for CD15, CD30, PAX- 5 (weakly) and negative for CD3, CD20, CD45, and CD79a.
We describe a unique case of classical HL that is CD30- positive but CD15- negative limited only to the liver without lymphadenopathy presenting as fever of unknown origin, pancytopenia and acute hepatic failure.
Case Report
A 63-year-old male from Bangladesh presented with a three-week history of persistent fevers, periumbilical abdominal pain, dysuria and jaundice. On physical exam, the vital signs were pertinent for tachycardia (HR 115bpm) and fever (T 39.4°C). The patient was noted to have elevated jugular venous pressure, jaundice, icterus, right upper quadrant tenderness and hepatosplenomegaly. There was no ascites, palmar erythema, spider angiomata, or asterixis. Initial serological tests included: white blood cell count 3200/mcl (normal range 4000-1150/mcl), hemoglobin 10.5g/dL (12-16.3g/dL), platelets 44,000/mcl (150,000- 400,000), sodium 126mmol/L, liver function (ALT 159units/L, AST 200units/L), alkaline phosphatase 695 units/L, albumin 3.2g/dL, total protein 6.4g/dL, direct bilirubin 5.23mg/dL (total bilirubin 8.1mg/dL) and INR 1.3.
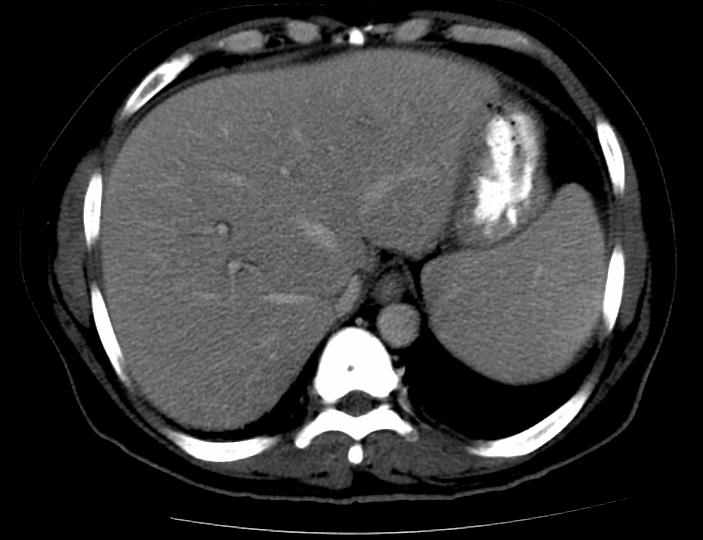
Abdominal ultrasound revealed a 1cm calcified granuloma in the right lobe of the liver with no intrahepatic or extrahepatic biliary ductal dilatation. An abdominal CT scan revealed an enlarged liver with normal contour and attenuation as well as a calcified granuloma in the right hepatic lobe. No discrete liver mases, intrahepatic or extrahepatic biliary ductal dilatation were identified. The spleen was mildly enlarged, measuring 18.5 cm without focal lesions (Figure 1). An MRCP was ordered to further evaluate the biliary tract and showed no intrahepatic or extrahepatic biliary tract dilatation. A CT scan of the chest was also performed, showing inflammatory changes in the region of the pancreatic head and first portion of the duodenum. Notably, there was no abnormal lymphadenopathy on the CT scans.
The patient’s clinical condition continued to worsen, as the intermittent fevers failed to resolve despite various antibiotic administrations. Infectious disease workup was found to be negative for the following: HSV, RPR/VDRL, HHV6, parvovirus B19, CMV, EBV, HIV, Hepatitis A, Hepatitis B, Hepatitis C, Hepatitis E, Leishmania, toxoplasmosis, schistosomiasis, babesia, coxiella burnetti, bartonella henselae, and legionella. The patient was empirically treated for Tuberculosis. A rheumatologic workup was negative for the following: smooth muscle antibody (Ab), anti-mitochondrial Ab, double-stranded DNA, IgG4, and ANA. Complement levels were within normal limits. All blood, urine and stool cultures remained negative throughout the hospital course.
The patient underwent a bone marrow aspiration and biopsy for continued fevers and pancytopenia. Pathology revealed a moderately hypercellular bone marrow with trilineage hematopoiesis, erythroid and megakaryocytic hyperplasia. There was no evidence of granulomata or of a lymphoid aggregate, acid fast bacilli or fungal elements. The differential cell count was normal. Flow cytometric evaluation was negative for lymphomatous or leukemic involvement of the bone marrow.
With continued rise in bilirubin and deteriorating clinical status, the patient underwent liver biopsy. The pathology revealed a liver parenchyma with nodular expansion of the portal and periportal areas by a mixed population of lymphocytes, eosinophils, histiocytes and large atypical lymphoid cells. Immunostaining was positive for CD30, Pax-5 (focal and weak), MUM-1, PDL-1, while negative for CD15, CD20, CD79a, OCT2, and BOB-1. Overall findings were consistent with liver parenchymal involvement by a CD30 lymphoproliferative disorder, favoring classical Hodgkin’s lymphoma involving the liver (Figures 2a-2c).
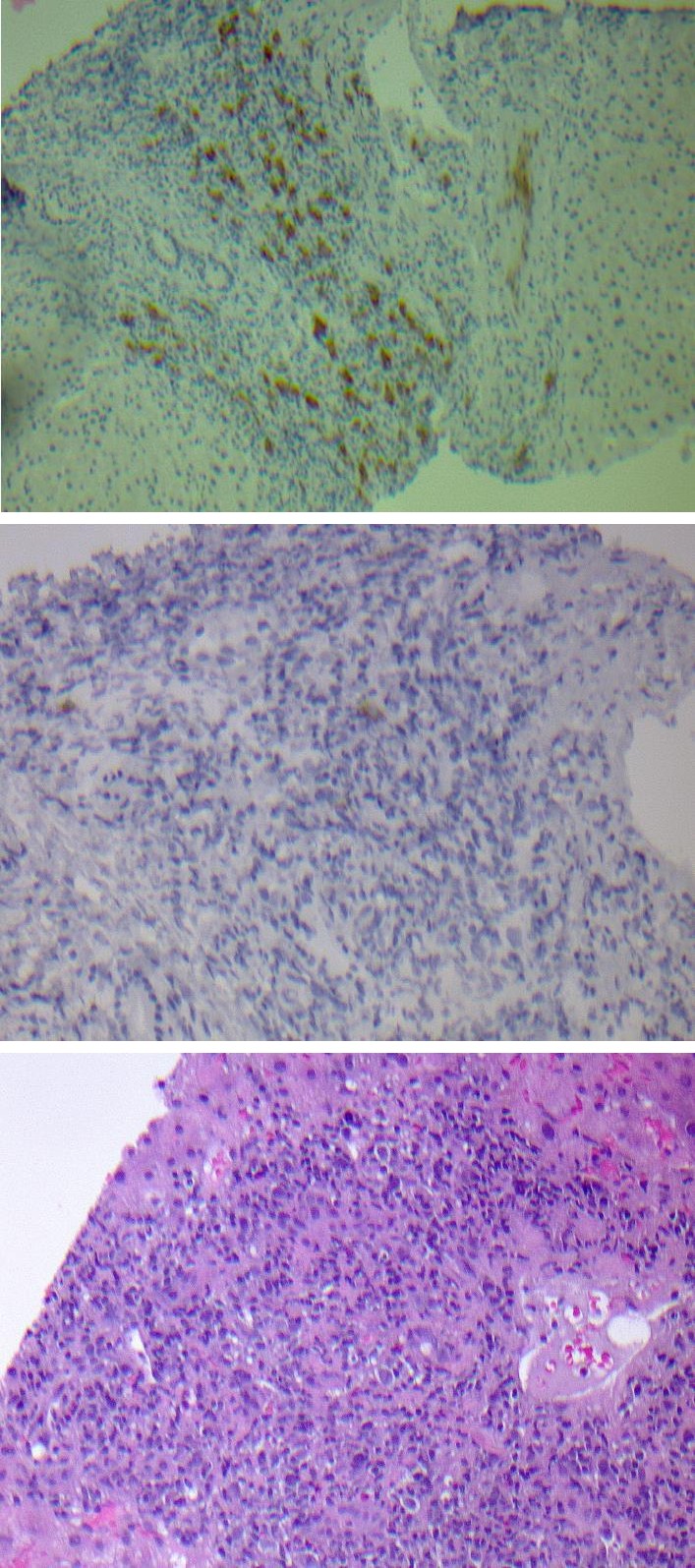
Figure 2a: Liver biopsy showing marked inflammation within a portal triad along with atypical large mononuclear cells displaying eosinophilic prominent nucleoli with an acute inflammatory background.
Figure 2b: Immunostain for CD30 was positive in atypical large mononuclear cells within the portal triads.
Figure 2c: Immunostain for CD15 was negative in atypical large mononuclear cells within the portal triads. Immunostains for viral markers including CMV and HSV were negative.
We administered pulse steroids with hydrocortisone in an effort to salvage the patient. A PET/CT scan was desired for complete staging but could not be obtained secondary to worsening clinical status. Chemotherapy could not be administered secondary to end stage liver failure. Despite all efforts, the patient developed acute respiratory failure and subsequently expired.
Discussion
Primary hepatic lymphoma (PHL) is typically defined as sole liver involvement without systemic lymphadenopathy. 20% of patients with HL will have hepatic involvement; however, it is unclear how many of those patients have only hepatic involvement [3]. The criteria for diagnosis of PHL as defined by Lei are the following: i) Symptoms caused by liver involvement at presentation; ii) Absence of distant liver involvement at presentation based on clinical and radiographic examinations and iii) Absence of blood involvement on a peripheral blood smears [4].
PHL represents about 0.016% of all Non-Hodgkin’s Lymphoma (NHL) and 0.4% of all extranodal lymphomas [5]. The most common histology of PHL is diffuse large B- cell lymphoma (DLBCL), occurring in approximately 60% of cases [6]. While most cases of PHL are of NHL histology, PHL of a HL histology is even rarer, scarcely cited within the literature. In a single center experience, 1662 liver biopsies were performed at the Glasgow Royal Infirmary, 63 of which were found to have hepatic granulomas, and only 6.3% were found to have underlying HL [7]. Our case is unique in that the PHL is of classical HL histology, different from the common DLBCL histology.
Arriving at the diagnosis of HL can be challenging because of the i) small size of biopsy specimens, ii) the sparsity of diagnostic RS cells and other variants, and iii) association with nonspecific inflammatory changes which can delay arriving to the correct diagnosis. Dich, et al. [8] found that patients with hepatic involvement of HL were more likely to have evidence of portal infiltrates larger than 1mm in diameter, acute cholangitis, portal edema, and portal infiltrates with mostly atypical lymphocytes when compared to patients without hepatic involvement. In our case, what delayed obtaining a biopsy was the absence of any radiographic lesions in the liver to sample.
Common symptoms of patients with unrecognized HL include prolonged fever, weight loss and night sweats, experienced by 67%, 67%, and 19% respectively in one series [9]. Febrile cholestatic disease as an initial presentation of NLPHL has been reported [10]. Marinone, et al. [11] described two cases of HL confined to the liver and bone marrow without systemic lymph node involvement presenting as acute cholestatatic disease. Cervantes, et al. [12] studied 421 consecutive patients diagnosed with HL and found that 6 patients (1.4%) presented with liver abnormalities, 0.7% had no evidence of extrahepatic disease, cholestasis was observed in all six cases, four had jaundice, and one had hepatic failure. In one case presented by Zaman, et al. [13], the manifestation of HL in a 29-year-old male was with liver abscesses. Our patient presented with febrile acute cholestatic disease with jaundice, icterus, right upper quadrant pain, and hepatosplenomegaly.
PHL can manifest radiographically in different ways. PHL can present as a solitary discrete lesion, which is the most common presentation of PHL, but can also present as multifocal lesions, diffuse infiltration, or as a periportal mass [14]. What makes our case challenging was that the only abnormality on imaging of the liver was hepatomegaly on the CT abdomen and pelvis.
There is little consistency regarding the treatment and outcomes of PHL within the literature. Some clinicians have treated patients with PHL with surgery alone, while others have combined chemotherapy with surgery [15] and/or radiation [16]. In a series of 8 patients with NHL presenting as a primary tumor of the liver, 4 out of 8 patients who received either surgery alone or a combination of surgery and chemotherapy achieved a complete remission, while the remaining 4 patients died within 3 months of diagnosis [15]. A single case report described a patient with unresectable NHL of the liver who was treated with combination of chemotherapy and radiation who was found to be free of disease at 37 months [16]. There is one reported case in the pediatric population of a 7-year-old boy presenting with fulminant hepatic failure diagnosed with NLPHL who was treated with a liver transplant, relapsed and subsequently treated with chemotherapy, and was disease-free at 1-year follow-up [17]. Patients with primary hepatic Hodgkin’s lymphoma, especially presenting with febrile cholestatic disease, are associated with more clinically and histologically aggressive types of HL including mixed cellularity and lymphocyte depletion [18].
In our case, the absence of a definable lesion in the liver and systemic lymphadenopathy despite worsening transaminitis and direct hyperbilirubinemia made obtaining a diagnosis difficult. It is important to evaluate and exclude other conditions which can mimic HL. Biopsy of the liver revealed nodular expansion of the portal and periportal areas by a mixed population of small mature lymphocytes, eosinophils, histiocytes and large atypical lymphoid cells. The immunohistochemical staining was positive for CD30 and focally weak positive for Pax-5 and negative for the following: CD15, CD20, CD79a, CD3, CD2, ALK, granzyme B, Bcl-2, Bcl-6, OCT-2, BOB-1, CMV, HSV I
and II, and FISH for EBV. This combination of immunoreactivity ruled out EBV-associated large cell lymphoma and anaplastic large cell lymphoma and favoured involvement of the liver parenchyma by a CD30- positive lymphoproliferative disorder, favoring classical HL. Ideally, a PET/CT scan should have been ordered to evaluate the extent of disease and to help determine involvement of the spleen by HL especially in light of the splenomegaly present on CT scans. However, due to the patient’s worsening clinical condition once the biopsy results were obtained, a PET/CT scan could not be obtained. Although the possibility of splenic involvement by lymphoma does exist, we believe that the mild splenomegaly was sequelae from acute liver failure.
In addition, HLs typically are positive for CD15 and CD30 expression, whereas our patient was CD30-positive and CD15-negative. This unique phenotype has been reported in the literature to be a variant of HL; Konkay, et al. [19] showed that 52 of 155 (33.5%) patients diagnosed with HL who had their immunohistochemistry profiles analyzed were CD30-positive and CD15-negative. Von Wasieleski, et al. [20] evaluated 1751 cases of HL and found that 12% lacked CD15 positivity and showed that CD15-negative expression was an independent negative prognostic factor for relapse and survival. Although it is known that the CD30-positive CD15-negative phenotype can be found in HL, the manifestation of this disease as a primary hepatic lymphoma without any other systemic lymphadenopathy, to our knowledge, has not been reported in the literature. Arriving at a clear diagnosis can be difficult but is usually made with a combination of morphological description of the architecture, tumor immunophenotype, background phenotype and genotype.
Conclusion
In conclusion, primary hepatic lymphoma should be considered in a patient who presents with unexplained fever, weight loss, and cholestasis. This case was unique in its clinical presentation, solitary organ involvement and uncommon immunophenotype of CD30-positivity and CD15-negativity. If there is no radiographic identifiable lesion to biopsy, random sampling of the liver should be performed if the etiology of liver failure is unknown. Though exceedingly rare, Hodgkin’s lymphoma limited to the liver requires prompt identification and treatment to allow for the best possible outcome. Conflicts of Interest: The authors declare that there is no conflict of interest regarding the publication of this article.
References
-
SEER Cancer Stat Facts: Hodgkin Lymphoma. National Cancer Institute. Bethesda, MD.
-
Carbone PP, Kaplan HS, Musshoff K, Smithers DW, Tubiana M (1971) Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 31(11): 1860-1861.
-
Ross A, Friedman LS (2006) The liver in systemic disease. In: Bacon BR, (Eds.), Comprehensive clinical hepatology, 2nd edn. Mosby Elsevier Ltd, Philadelphia, pp: 537-547.
-
Lei KI (1998) Primary non-Hodgkins lymphoma of the liver. Leuk Lymphoma 29(3-4): 293-299.
-
Loddenkemper C, Longerich T (2008) Lymphoma of the liver. In: Cavalli F, (Eds.), Extranodal Lymphomas, Pathology and Management. Abingdon, UK: CRC Press, pp: 277-288.
-
Ugurluer G, Miller RC, Li Y, Thariat J, Ghadjar P, et al. (2016) Primary Hepatic Lymphoma: A Retrospective, Multicenter Rare Cancer Network Study. Rare Tumors 8(3): 6502.
-
Gaya DR, Thorburn D, Oien KA, Morris AJ, Stanley AJ (2003) Hepatic granulomas: a 10 year single centre experience. J Clin Pathol 56(11): 850-853.
-
Dich NH, Goodman ZD, Klein MA (1989) Hepatic involvement in Hodgkin's disease. Clues to histologic diagnosis. Cancer 64(10): 2121-2126.
-
Hasle H, Mellemgaard A (1993) Hodgkin's disease diagnosed post mortem: a population based study. Br J Cancer 67(1): 185-189.
-
Mrzljak A, Gasparov S, Kardum-Skelin I, Colic-Cvrlje V, Ostojic-Kolonic S (2010) Febrile cholestatic disease as an initial presentation of nodular lymphocyte- predominant Hodgkin lymphoma 16(35): 4491-4493.
-
Marinone G, Lazzari R, Pellizzari F, Marinone MG (1989) Acute cholestatic Hodgkin's lymphoma: an unusual clinical picture. Haematologica 74(3): 293- 296.
-
Cervantes F, Briones J, Bruguera M, Font C, Grau JM, et al. (1996) Hodgkin's disease presenting as a cholestatic febrile illness: incidence and main characteristics in a series of 421 patients. Ann Hematol 72(6): 357-360.
-
Zaman A, Bramley PN, Wyatt J, Simmons A, Losowsky MS, et al. (1991) Hodgkin’s disease presenting as liver abscesses. Gut 32(8): 959-962.
-
Rajesh S, Bansal K, Sureka B, Patidar Y, Bihari C, et al. (2015) The imaging conundrum of hepatic lymphoma revisited. Insights Imaging 6(6): 679-692.
-
Scoazec JY, Degott C, Brousse N, Barge J, Molas G, et al. (1991) Non- Hodgkin’s lymphoma presenting as a primary tumor of the liver: presentation, diagnosis and outcome in eight patients. Hepatology 13(5): 870-875.
-
Czerniak A, Soreide O, Halnan K, Krausz T, Edwards WH, et al. (1987) Primary non-Hodgkin’s lymphoma of the liver. Eur J Surg Oncol 13: 251-255.
-
Woolf KM, Wei MC, Link MP, Arber DA, Warnke RA (2008) Nodular lymphocytepredominant Hodgkin lymphoma presenting as fulminant hepatic failure in a pediatric patient: a case report with pathologic, immunophenotypic, and molecular findings. Appl Immunohistochem Mol Morphol 16(2): 196-201.
-
Jaffe ES (1987) Malignant lymphomas: pathology of hepatic involvement. Semin Liver Dis 7(3): 257-268.
-
Konkay K, Paul TR, Uppin SG, Rao DR (2016) Hodgkin lymphoma: A clinicopathological and immunophenotypic study. Indian J Med Paediatr Oncol 37(1): 59-65.
-
Von Wasielewski R, Mengel M, Fischer R, Hansmann ML, Hübner K, et al. (1997) Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am J Pathol 151(4): 1123-1130.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies