Diversity of Clinical Features of Patients with Thalassemia Intermedia
Thalassemia is the most common single gene disorder in the world found at high frequencies in many populations worldwide. Beta thalassemia presents with extremely diverse phenotypes within the homozygous and compound heterozygote states. The term “β-thalassemia intermedia†(TI) was first suggested to describe patients who had clinical manifestations that are not as mild as β-thalassemia minor and not as severe as β-thalassemia major†(TM). Patients with TI usually present to medical attention in later childhood or even adulthood. Diagnosis of thalassemia intermedia relies on clinical presentation more than the molecular characterization. In this study clinical features of the patients having thalassemia intermedia were observed. The study was carried out on 100 known thalassemic intermedia patients. All of the patients were diagnosed clinically and confirmed with molecular analysis. The clinical features observed age at the time of examination, age at the start of transfusion, interval between transfusion, facial changes, spleen and liver. Presence of jaundice in the patients was also noted. The ages of the patients were between 2 to 34 years. The age at commencement of transfusion was between 1 year to 32 years. 21 patients had severe facial changes, 48 with mild facial changes while 31 had no facial changes. Only 6 patients had enlarged spleen. Six of the patients had their spleen removed. 5 patients showed up with significant jaundice and 38 patients had enlarged liver. Patients presented with broad spectrum of clinical features. Majority of the patient had mild changes in liver and spleen, having mild facial changes and less sever jaundice. While others represented with sever hemolysis , enlarged spleen and liver accompanied with significant facial changes and yellow coloration of skin representing jaundice. Thus it is difficult a draw a definite line between thalassemia intermedia and thalassemia major. A careful observation of physical presentation and molecular analysis support is required to claim the diagnosis of thalassemia intermedia.
Introduction
Thalassemia is the most common single gene disorder in the world found at high frequencies in many populations worldwide. Beta thalassemia presents with extremely diverse phenotypes within the homozygous and compound heterozygote states. The terms thalassemia major (TM) and thalassemia intermedia (TI) encompass a wide spectrum of clinical, as well as laboratory abnormalities [1]. The term “β-thalassemia intermedia” (TI) was first suggested to describe patients who had clinical manifestations that are not as mild as β-thalassemia minor and not as severe as β-thalassemia major” (TM). Patients with TI usually present to medical attention in later childhood or even adulthood [2]. Diagnosis of thalassemia intermedia relies on clinical presentation more than the molecular characterization. In this study the clinical features of the patients having thalassemia intermedia were observed.
Materials and Methods
The study was carried out on 100 known thalassemic intermedia patients. The criteria followed for the inclusion of the patients was;
- Patient was known thalassemic
- Age at commencement of transfusion was more than three years
- The interval between the transfusion was at least 1 month All the patients were diagnosed clinically and confirmed with molecular analysis. The clinical features of the patients including age at the time of examination, age at the start of transfusion, interval between transfusion, facial changes, spleen and liver were observed. Presence of jaundice in the patients was also noted.
Results
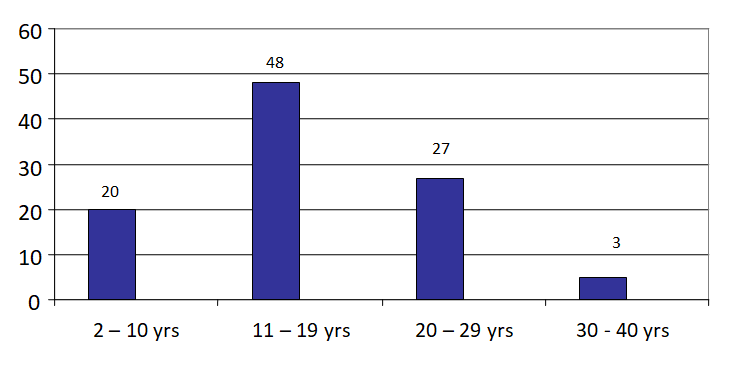
The patients were distributed in four different grades, Severe, Moderated, Mild and very -mild depending on the start of transfusion. Out of the 100 patients selected for the study 53 belonged to Severe TI, 37 were classified as Moderate TI, 7 were in Mild TI group and 3 were included in Very mild type of thalassemia intermedia patients (Table 1). The CI for the grades was 1.495-1.704 and the P value was P =0.00001. The age of these patients were between 2–34 years, majority being between 11-19 Years (Figure 1). In this group of patients 49 were females and 51 were males (Table 1).
The ages of the patients were between 2 to 34 years. The age at commencement of transfusion was between 1 year to 32 years. 21 patients had significant facial changes, mild facial changes were seen in 48 patients and 31 with no facial changes. Only 6 patients had enlarged spleen. Six of the patients had their spleen removed. 5 patients showed up with sever jaundice and 38 patients had enlarged liver Table 2. The clinical features found in these patients including facial changes, spleen, liver and jaundice are given in the Table 2.
| Grades | Frequency | Percent | Cumulative Percent |
|---|---|---|---|
| Severe | 53 | 53 | 53 |
| Moderate | 37 | 37 | 90 |
| Mild | 7 | 7 | 97 |
| Very Mild | 3 | 3 | 100 |
| Total | 100 | 100 |
Table 1: Grades of thalassemia Intermedia.
| Features | Grades of TI | |||
|---|---|---|---|---|
| I | II | III | IV | |
| Male | 28 | 16 | 5 | 2 |
| Females | 25 | 21 | 2 | 1 |
| Mean age at the time of examination | ||||
| 13.7 yrs (2 – 30 yrs) | 17.8 yrs (5 – 27 yrs ) | 24.4 yrs (12 - 36 yrs) | 30 yrs (24 – 34 yrs) | |
| Mean age at the start of transfusion | ||||
| 3.4 Years (3 months – 28 years ) | 6.3 yrs (4 – 10 yrs) | 12 years (11- 18years) | 28 years ( 24 – 32 yrs) | |
| Interval between Transfusion | ||||
| 2 or > 2 /year | 51 | 36 | 7 | 3 |
| 1 or > 1 / year | 2 | 1 | - | - |
| Facial changes | ||||
| n | 18 | 10 | 2 | 1 |
| m | 25 | 17 | 4 | 2 |
| s | 10 | 10 | 1 | - |
| Spleen | ||||
| n | 21 | 16 | 3 | 2 |
| y | 3 | 1 | 1 | 1 |
| Splenectomy | ||||
| n | 50 | 36 | 6 | 2 |
| y | 3 | 1 | 1 | 1 |
| Jaundice | ||||
| 0 | 11 | 5 | 1 | - |
| 1 | 12 | 10 | 1 | - |
| 2 | 27 | 21 | 4 | 3 |
| 3 | 3 | 1 | 1 | - |
| Liver | ||||
| n | 22 | 15 | 3 | 2 |
| e | 31 | 22 | 4 | 1 |
Table 2: Clinical features of Thalassemia Intermedia patients.
Grads of TI: I = Severe TI, II = Moderate TI, III = Mild TI, IV= Very mild TI. Liver: n = Normal, e = Enlarged Jaundice: 0 = Absent, 1 = Mild, 2 = Moderate, 3 = Severe. Splenectomy: y = Yes, n = No Faces: n = Normal, m = Mild changes, s = Significant changes Table 2: Clinical features of Thalassemia Intermedia patients.
Discussion
Beta thalassemis are characterized by imbalance of chain synthesis, the excess of free globin chains accumulates within erythroid cells. The chains are aggregated denatured and degraded. Degradation of the chains results in the formation of insoluble precipitates and hemichromes that damages the cell membranes leading to ineffective erythropoiesis in the bone marrow. Intravascular hemolysis and binding of antibodies and complement to the red cells triggers hemolysis of red cells in spleen. This hemolysis leads to diminished tissue oxygenation increasing the levels of erythropoietin stimulating the bone marrow. Thus the expansion of bone marrow causes skeletal deformities and osteopenia. The degradation of red cells causes increase iron absorption which further contribute to iron overload [3]. Since the hallmark of disease in these syndromes is ineffective erythropoiesis, peripheral hemolysis, and subsequent anemia, transfusion- dependence has been an essential factor in characterizing the various thalassemia phenotypes and their severity [4].
Beta Thalassemia presents in three forms thalassemia major, thalassemia intermedia and thalassemia minor. The different phenotypes of beta thalassemia reckons on the clinical severity of the disease. Thalassemia intermedia usually demonstrate mild to moderate anemia and a hemoglobin level ranging between 7 and 10 g/dL, which is sustainable without the need for regular transfusion therapy. Unfortunately, some TI patients are set on regular transfusion particularly unnecessary transfusion are given to the patients of TI if they present during a period of intercurrent infection requiring a few transfusions [3]. According to the results mean age for the initiation of transfusion was 3 years in the grade I and 28 years in the grade IV. Majority of the patients were transfusion independent having the interval between the transfusion of 2 or more years. However one of the patient needed transfusion at the age of 3 months, the patient was identified with Cap+1 mutation and later in life the patient being non-transfusion dependent received transfusion with an interval of one year or more. If transfusion therapy is initiated during the first years of life and if the Hb is maintained at 9-10 g/dl these manifestations are usually absent or minimally present in thalassemia major. Transfusion for thalassemia intermedia vary considerably [2].
Majority of the patients in this study had no or mild Facial changes. 21 patients came up with sever changes on their faces including frontal bossing depressed nasal bridge, and maxillary protrusion secondary to marrow expansion. Usually, these manifestations are absent or minimally present in patients with thalassemia major if transfusion therapy is initiated early during the first year of life provided that the hemoglobin levels are maintained at 9–10 g/dL. Where transfusion is readily available, the classical physical findings of thalassemia are now more commonly found in patients with thalassemia intermedia, particularly those who are at the severe end of the phenotypical spectrum. In such patients, the physical manifestations can be suppressed in part with subsequent transfusion [2].
Abdominal enlargement due to hepatosplenomegaly was observed in 6 patients and 6 patients had undergone splenectomy due to large spleen size and increasing anemia and increasing transfusion requirement. The natural history of thalassemia intermedia is highly variable, complications of severe anemia are common. The need of splenectomy has been indicated due to large spleen size, with increasing anemia or increasing transfusion requirement with or without a decrease in neutrophil or platelet count5. The hallmark of thalassaemia syndromes is the production of defective red blood cells that are removed by the spleen resulting in an enlarged hyperfunctioning spleen (splenomegaly).Removal of the spleen may thus prolong red blood cell survival by reducing the amount of red blood cells removed from circulation and may ultimately result in the reduced need for blood transfusions [5].
Splenectomy in TI had been considered a significant risk factor for many disease-related complications, in particular, thrombosis and postmenopausal hormone therapy (PHT). Furthermore, chronic thromboembolism in splenectomized TI patients was linked with a high frequency of PHT. In light of the above morbidities associated with splenectomy and despite the advantage of splenectomy in maintaining higher Hb levels, clinical practice is gradually shifting to restrict indications, growth retardation, hypersplenism with symptomatic leukopenia, and/or thrombocytopenia or symptomatic hypersplenism [6].
Majority of the pateints had only mild to moderate jaundice and only five patients showed yellow discoloration of skin. Thalassemia intermedia patients with Hb of much below 7 or 8 gm/dl and excess energy consumption due to the profound hemolysis can produce small stature, poor weight gain, poor energy levels, susceptibility to infection and yellow discoloration (jaundice) of the skin, eyes, and mucous membranes caused by increased amount of bilirubin in the blood [7, 1]. The different phenotypes of beta thalassemia reckons on the clinical severity of the disease. Thalassemia intermedia usually demonstrate mild to moderate anemia and a hemoglobin level ranging between 7 and 10 g/dL, which is sustainable without the need for regular transfusion therapy. Unfortunately, some TI patients are set on regular transfusion particularly unnecessary transfusion are given to the patients of TI if they present during a period of intercurrent infection requiring a few transfusions [3].
Conclusion
Thalassemia intermedia may present with a broad d spectrum of clinical features. Majority of the patient had mild changes in liver and spleen, having mild facial changes and less sever jaundice. Some of the patients having mild clinical features still required transfusion. While on the other hand some of the patient represented with features of sever form of the disease. These patients had features of sever hemolysis, enlarged spleen and liver accompanied with significant facial changes and yellow coloration of skin representing jaundice. Thus it is difficult to draw a definite line between thalassemia intermedia and thalassemia major. A careful observation of physical presentation and molecular analysis support is required to claim the diagnosis of thalassemia intermedia. Among the features to be noted are age of the initiation of transfusion and non-dependence of the patient on transfusion for survival. A significant support for the diagnosis is the molecular analysis that include the presence of mild mutations, presence of ameliorating factors like Xmn-I polymorphism, coexistence of alpha thalassemia and other factors manipulating the production of hemoglobin F. Thus a clinical assessment requires the support of molecular analysis to depict the complete picture of thalassemia intermedia.
Acknowledgement
I would like to extend my deepest gratitude to Gen. Suhaib Ahmed of Armed forces institute of pathology, Rawalpindi for his marvelous supervision in the study. Thanks are also due for the technical staff of the institute for their tremendous support. The research work was partially funded by baqai institute of hematology, PNS shifa Karachi, Armed forces institute of hematology Rawalpindi plus including some self funds.
References
-
Haidar R, Mhaidli H, Taher AT (2010) Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J 19(6): 871-878.
-
Nienhuis AW, Nathan DG (2012) Pathophysiology and Clinical Manifestations of the β-Thalassemias. Cold Spring Harb Perspect Med 2(12): a011726.
-
Musallam KM, Taher AT, Rachmilewitz EA (2012) β-Thalassemia Intermedia: A Clinical Perspective. Cold Spring Harb Perspect Med 2(7): a013482.
-
Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA (2013) Non-transfusion-dependent thalassemias. Haematologica 98(6): 833-844.
-
Sharma A, Mathew ME, Aravindakshan R (2019) Splenectomy for people with thalassaemia major or intermedia. Cochrane Systematic Review.
-
Amin SS, Jalal SD, Ali KM, Mohammed AI, Rasool LK, et al. (2020) Beta-Thalassemia Intermedia: A Single Thalassemia Center Experience from Northeastern Iraq. BioMed Research International 2020: 2807120.
-
Sharma DC, Arya A, Kishor P, Woike P, Bindal J, et al. (2017) Overview On Thalassemias: A Review Article. Med Res Chron 4(3): 325-337.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies