Evan’s Syndrome - A Case Report
Evan’s syndrome is a rare and chronic autoimmune disease characterised by autoimmune hemolytic anemia and immune thrombocytopenic purpura with a positive Coomb’s test in the absence of an underlying etiology. Majority of the patients with this syndrome have a chronic relapsing course and significant morbidity and mortality despite treatment. We present a case of a 22 year old female, in which, based on the clinical features, Coombs test, hemolytic anemia and thrombocytopenia, a diagnosis of Evans syndrome was made.
Introduction
Evans syndrome is an uncommon, complex chronic autoimmune disease which is characterized by autoimmune hemolytic anemia and immune thrombocytopenic purpura with a positive coombs test in the absence of an underlying etiology. This syndrome shows a variable complicated clinical course with remissions, exacerbations and association with significant morbidity and mortality [1]. Worldwide, it is diagnosed in less than 5% of all patients with either ITP or AIHA at onset. There are very few reports of Evans syndrome from the Indian subcontinent hence the incidence in India is relatively unknown.
Case Report
A 22 year old female reported with complaints of fatigue, fever, generalized abdominal pain and hematuria for about 1 week. She had similar episodes for last 1 year for which she was treated. There were recurrent exacerbations and she was referred to a tertiary care center for further investigations. Diagnosis of Evans syndrome was made on the basis of medical history, physical examination and laboratory investigations. Clinical features were those of anemia such as pallor, fatigue and dizziness as well as signs of thrombocytopenia including purpuras, hematuria, menorrhagia and mucosal bleeding. The presence of jaundice indicated hemolysis.
Hemogram showed hemoglobin of 2.1 gm/dl, total leucocyte count of 26000/cumm, differential leucocyte count- polymorphs 55%, lymphocytes 36%, eosinophils 5%, monocytes 4% and platelets showing severe thrombocytopenia with platelet count being 15000/cumm. On peripheral blood examination the smear showed Macrocytic Normochromic picture along with Macro-ovalocytes. There was agglutination of red blood cells along with presence of spherocytes, schistocytes, polychromasia and nucleated red blood cells. Reticulocytosis with reticulocyte count of 30% was seen.
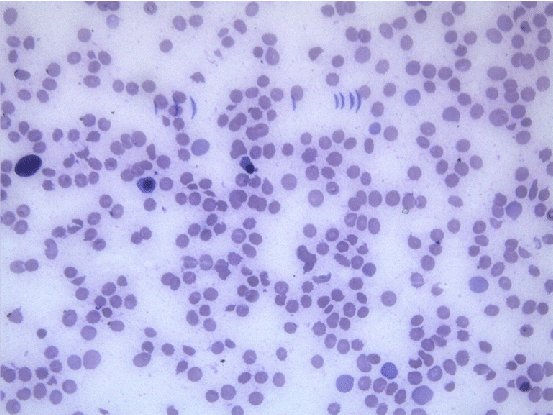
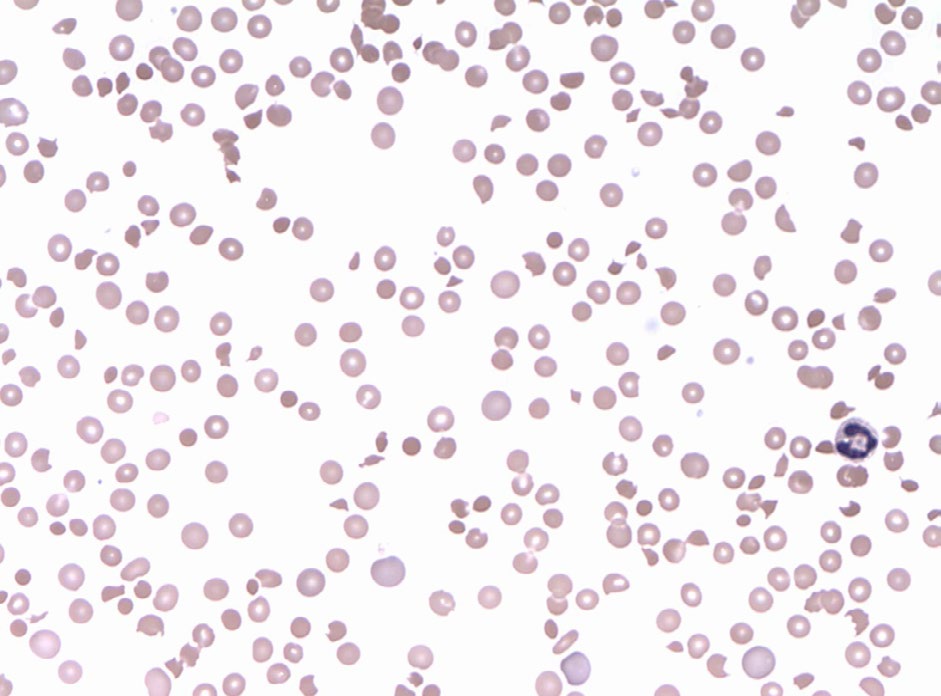
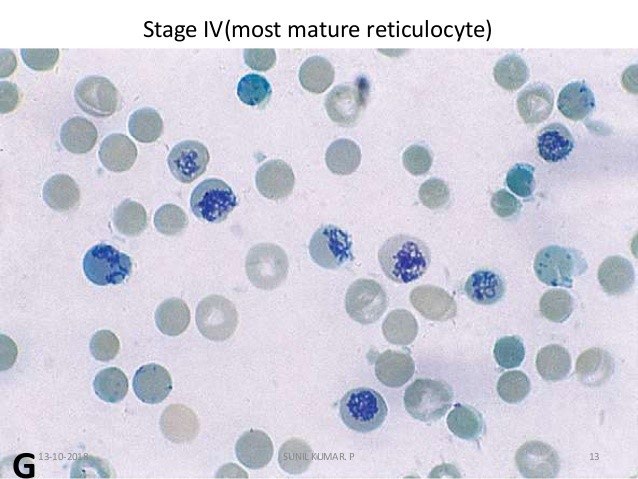
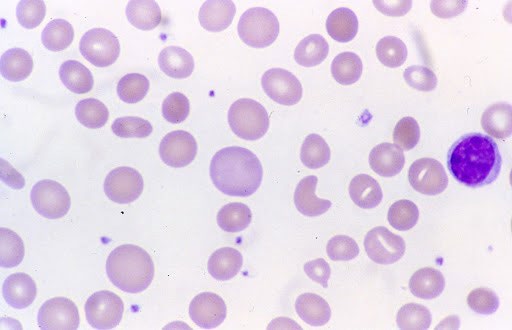
Direct antiglobulin (Coomb’s) test was advised and found to be positive. Total serum bilirubin was 8.1 mg/dl. Urinalysis showed dark yellow urine and negative for bilirubin. Evans syndrome is a diagnosis of exclusion and confounding factors such as infections, malignancies and rheumatological diseases should be ruled out. Test for lupus antibody was negative. ELISA for HIV I and II was found to be non-reactive. Viral serology was negative. Malarial antigen test was also negative. Chest X-ray and Abdominal ultrasonography was normal. There were no coincidental or precipitating infections or immune disorders. Thus, the patient, at the time of presentation showed the features of autoimmune hemolytic anemia, autoimmune thrombocytopenia and neutropenia with no evidence of infection, malignancy or any other obvious underlying etiology.
Discussion
Autoimmune hemolytic anemia (AIHA) is an immune disorder which is characterised by circulating antibodies against antigens on the red blood cells (RBCs) membrane resulting in shortened life span of the RBC [2]. Robert Evans first described an association between idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia in 1951. It was characterized by simultaneous destruction of the body’s own red blood cells, white blood cells, platelets, neutrophils which causes Autoimmune Hemolytic Anemia (AIHA) and Idiopathic Thrombocytopenia Purpura (ITP) or immune neutropenia in absence of any cause [3].
Evans syndrome is predominantly a disease of pediatric age group [4] though it has been reported in older individuals as also seen in our case. Among adults, Evans syndrome is known to affect women more often than men [5]. As Evans
syndrome is a diagnosis of exclusion so other confounding factors such as malignancies, infections and rheumatological disorders should be ruled out. A bone marrow examination (aspiration/biopsy) although necessary to rule out causes of aplastic anemia or an infiltrative disorder; however is not essential in classical cases of Evans syndrome when patients present with autoimmune hemolytic anemia or immune thrombocytopenia. Hence bone marrow aspiration was not required in our case.
Bone marrow aspiration when done usually shows a mild to moderate erythroid hyperplasia. Megakaryocytes may be normal to increased in number which indicates an increased destruction of platelets in the peripheral blood as the cause of thrombocytopenia [6]. There have been few cases reported of drug induced Evans syndrome associated with the use of diclofenac and ramipril. In our case, there was no history of such or any other medication [7]. The aetiology of Evans syndrome is reported to be due to an underlying deficiency in humoral or cell mediated immunity. Isolated episodes of thrombocytopenia and haemolytic anaemia and the results of in vitro studies have suggested that noncross reacting autoantibodies are targeted at different antigenic determinants on red cells and platelets.
Evans syndrome is managed by Corticosteroids and/ or intravenous immunoglobulins as the first-line therapy. Most patients respond to this line of treatment although relapses are quite common. The second line therapy includes immunosuppressive drug. Recently, some patients have been treated with rituximab. Rituximab is one such drug that can be tried in cases of refractory Evans syndrome. Rituximab is a chimeric anti-CD20 monoclonal antibody with human IgG1 and k constant regions and murine variable regions [8, 9]. It causes selective depletion of B cells through complement and antibody-dependent cell-mediated cytotoxicity and through induction of apoptosis.
Splenectomy may also be considered although long-term remissions are less frequent than in uncomplicated ITP. For severe and refractory cases, stem cell transplantation (SCT) offers the only chance of long-term cure. A fully developed Evans syndrome should be associated with a Coomb’s positive hemolytic anemia which is essential for definitive diagnosis. Although rare, Evans syndrome should be suspected and investigated for in patients presenting with autoimmune haemolytic anaemia or autoimmune thrombocytopenia concurrently or sequentially. Patients of either of these disorders individually should therefore be thoroughly investigated for the other so as not to miss a diagnosis of Evans syndrome. Our patient has yet not reached remission. The patient continues to have clinically evident anemia and thrombocytopenia despite steroid therapy. Hence, long term follow up is essential in such cases as the patient is at a risk of developing other autoimmune problems [4].
Conclusion
Although a rare disorder, Evans syndrome should always be considered in cases presenting with AIHA or AITP occurring simultaneously or in follow up after excluding the causes of unknown etiology. Hence the early diagnosis, knowledge of its presentation and a constant follow up of this syndrome is crucial.
References
-
Norton A, Roberts I (2006) Management of Evans syndrome. Br J Haematol 132: 125-137.
-
Agarwal B (1998) Autoimmune hemolytic anemia. Indian J Pediatr 65: 663-668.
-
Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, et al. (2009) The spectrum of Evan’s Syndrome in adults; new insight into the disease based on the analysis of 68 cases. Blood 114(15): 3167-3172.
-
Wang WC (1988) Evans syndrome in childhood: pathophysiology, clinical course and treatment. Am J Pediatr Hematol Oncol 10(4): 330-338.
-
Pui CH, Wilimas J, Wang W (2006) Evans syndrome in childhood. Eur Haematol 97(5): 754-758.
-
Savasan S, Warrier I, Ravindranath I (1997) The spectrum of Evans’ syndrome. Arch Dis Child 77(3): 245-248.
-
Keung YK, Mallarino MC, Cobos E (1998) Drug-induced evans syndrome. Ann Intern Med 128(4): 327.
-
Miller BA, Schultz Beardsley D (1983) Autoimmune pancytopenia of childhood associated with multisystem disease manifestations. J Pediatr 103(6): 877-878.
-
Grillo Lopez AJ, White CA, Varns C, Shen D, Wei A, et al. (1999) Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin Oncol 26: 66-73.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies