Novel Genetic Variants of the PROS1 Protein S Gene of an Individual with Protein S Deficiency
Introduction: Human Protein S (PS) is an anticoagulant essential for maintaining hemostasis. Although rare, PS deficiency can cause life-threatening thrombophilia. We found novel genetic variations in the PROS1 gene of a 46-year-old woman who was diagnosed with PS deficiency. We hypothesize how the mutations affect PS level and function. Materials and Methods: Citrated plasma was isolated from the patient’s blood. Biochemical analyses were conducted via activated partial thromboplastin time and thrombin generation assays with the addition of various concentrations of PS. Immunoblotting and ELISA were used to measure levels of PS in the woman’s plasma. We sequenced the entire PROS1 gene on chromosome 3 for two control samples and the individual’s sample. Results: We found two novel deletions and a novel insertion in the PROS1 gene of the patient. The deletions and insertion were predicted to activate cryptic splice acceptor and donor splice sites, respectively. In addition, the patient had alteration in whole blood coagulation parameters, including a prolonged aPTT and decreased plasma PS level. Conclusion: The genetic alterations in the patient’s PROS1 gene are predicted to cause inappropriate splicing of PS pre-mRNA that likely contributes to the Type I PS deficiency and abnormal coagulation properties of the study patient.
Abbreviations
DIC: Disseminated Intravascular Coagulation; aPTT: Activated Partial Thromboplastin Time; ETP: Endogenous Thrombin Potential; TFPI: Tissue Factor Pathway Inhibitor; APC: Activated Protein C.
Introduction
Our journey of understanding Protein S began in 1977 in Seattle, Washington, US, where the protein was first discovered and named after the city (S) [1]. Less than 10 years later in 1984, the physiological significance of Protein S (PS) as a natural anticoagulant was discovered when a patient experienced recurrent thrombosis due to a decreased level of PS [2]. Since then, our recognition of the several functions of PS, including as an anticoagulant, has advanced significantly.
Protein S is a 73-kDa vitamin K-dependent glycoprotein. Protein S circulates in plasma at total body concentration of 350 nM; hepatocytes contribute 40% of total secreted PS [3] and 5% of PS is reserved in platelet α-granules, which exclusively is derived from megakaryocyte expression [4]. Protein S contains several distinct domains, such as the thrombin-sensitive region and N-terminal Gla domain (among others), EGF1-4 domains and two sex hormone binding domains that are essential for mediating anticoagulation and other physiological functions of PS [5]. In plasma, 40% of PS circulates as free Protein S, and 60% of PS circulates bound to complement component 4 binding (C4BP). The free form of PS serves as the anticoagulant [6]; C4BP-bound PS has a non-hemostasis function in apoptosis [7].
Protein S is essential for hemostasis and regulation of the coagulation cascade [8]. Protein S is a cofactor for activated Protein C (APC); together, both PS and APC proteolyze FVIIIa and FVa and prevent the activation of Xase and prothrombinase, respectively [9]. Although APC alone can efficiently cleave FVa, the presence of PS enhances proteolysis by 20 to 30-fold [10]. Protein S also acts as a cofactor of tissue factor pathway inhibitor (TFPI), a single chain glycoprotein essential for inhibition of free factor Xa [11]. Together, TFPI and PS inhibit free FXa activity and the TF-FVIIa complex, thereby modulating the extrinsic pathway of coagulation [12]. Notably, PS also has a direct function in anticoagulation by binding and inhibiting FIXa, in the presence and absence of FVIIIa [13].
The essential functions of PS in anticoagulation are regulated at the level of expression of the PROS1 gene. Located on chromosome 3, the PROS1 gene is composed of 15 exons and 14 introns, including specific binding sites for transcription factors [14, 15]. Estrogen downregulates transcription of PROS1, and progesterone increases expression of PS by 25% [8, 16].
In addition to hormonal influences on PS regulation, external factors can promote acquired PS deficiency [17]. Kidney disease, estrogen-containing oral contraceptives, pregnancy, warfarin, and even cancer can decrease the level of PS, which heightens the risk of blood clotting and venous thromboembolism [18].
Unlike acquired PS deficiency, although rare, hereditary PS deficiency is an autosomal dominant condition in heterozygous individuals and is characterized by recurrent deep venous thromboembolism and pulmonary embolism [19]. Still rarer are cases of homozygous and heterozygous PS deficiency that cause the development of severe purpura fulminans during infancy, a complication of disseminated intravascular coagulation (DIC) [20, 21]. The prevalence of hereditary PS deficiency is unknown for the general population, and the incidence of this illness is thought to be relatively rare [22].
An elevated risk of thrombophilia due to irregular blood clotting is associated with PS deficiency and can lead to severe, life-threatening conditions, such as DIC, cerebrovascular hemorrhage, and recurrent venous thromboembolism [23]. To protect persons with hereditary PS-deficiency from these conditions, lifelong anticoagulation therapy is often necessary with low molecular weight heparin or direct oral anticoagulants [24]. However, prolonged use of these anticoagulants causes significant adverse effects, such as elevated bleeding risk [24].
Although over 400 mutations have been implicated in PS deficiency, these mutations fall under three categories of PS deficiency. The common type of inherited PS deficiency is Type 1, characterized by reduced levels of free and total PS and reduced function of PS [25]. In Type 2 PS deficiency, only PS function is reduced. Type 3 deficiency is defined by reduced free PS and functional PS with retention of normal overall PS levels [26, 27]. Mutations associated with Type 3 deficiency occur at a frequency of 1.65-1.8% in the Japanese population [27].
The patient discussed in this study is a Caucasian woman for whom a coagulation factor deficiency was suspected after her mother experienced recurrent thrombophilia and recurrent pregnancy loss. Further testing showed that the study patient had a reduced level of PS and reduced PS function, i.e., Type 1 PS deficiency. DNA sequencing showed that the individual had three novel mutations in the PROS1 gene, and biochemical assays revealed blood coagulation parameters. This study provides novel understanding of a rare, life-threatening disease that will guide future clinical treatment.
Material and Methods
Plasma Preparation
Blood was collected in 3.2% sodium citrate venipuncture collection tubes. Platelet poor plasma was prepared by centrifugation at 14,000 g for 20 minutes at 20°C and stored in aliquots at -80°C.
Thrombin Generation Assay
All reagents were prepared according to the manufacturer’s instructions. Technothrombin® TGA was from Diapharma. The height of thrombin peak, endogenous thrombin potential (ETP), and thrombin generation lag time were calculated with our standard curve and plotted with GraphPad Prism v.10 software. The thrombin generation assay was conducted with the addition of human PS at concentrations of 10 nM, 25 nM, 50 nM, 75 nM, and 150 nM.
Activated Partial Thromboplastin Time (aPTT)
All reagents and samples were prepared according to the manufacturer’s instructions with Technoclone Siron LIS® aPTT (5035119, DiaPharma). Absorbance of each sample was measured at 405 nm with a VERSA max microplate reader. Proteomic analyses were conducted via activated partial thromboplastin time (aPTT) with the addition of human PS at concentrations of 10 nM, 25 nM, 50 nM, 75 nM, and 150 nM.
Immunoblotting
Abundant serum proteins were removed from plasma by passing it through the High Select Depletion Spin Columns (Thermo Scientific™, USA). The Bradford assay was conducted to determine protein concentrations. Membranes were blocked with 5% non-fat dry milk overnight at 4°C. Protein S primary antibody (ab280885, Abcam) was used at 1:4000, and blots were further probed with goat anti-rabbit HRB-conjugated secondary antibody (ab6721, Abcam) at 1:4000 dilution. Transferrin was the control. The bands were detected using Pierce™ ECL Western Blotting Substrate (32209, Thermofisher Scientific) and analyzed with ImageJ software.
Next Generation Sequencing
Next-generation sequencing of the PROS1 gene was conducted with a Myseq system (Illumina). DNA was isolated with Purgegene Buccal Cell Core Kit (158845, Qiagen) according to manufacturer’s instructions, and the patient sample was compared with samples from two healthy controls. The PS primers used for sequencing PROS1 were as follows: 5′ - GTAAAACGACGGCCAG – 3′ and 5′ – CAGGAAACAGCTATGAC – 3′.
Splice Site Prediction
The effects of the novel variants on PROS1 mRNA splicing were predicted with Splice Site Prediction by Neural Network, MaxEntScan and Splice Sequences Finder Splice Site Analysis.
Enzyme Linked Immunosorbent Assay
An ELISA to measure PS in plasma samples was conducted according to the manufacturer instructions (ab125969, Abcam).
Statistical Analysis
Data were analyzed with one-way ANOVA and associated p-values with Bonferroni correction was performed with GraphPad Prism v.10 software (GraphPad Software, Inc.).
P-values <0.05 were considered statistically significant.
Results
Abnormal Blood Parameters of the Study Patient
Assay of the patient’s plasma by aPTT, peak thrombin generation, and ELISA for free PS demonstrated that the individual had Type I PS deficiency, defined as a decreased level of free PS and decreased activity of PS. The aPTT value of the PS-deficient individual was 28 s, compared with a normal reference range of 30 to 40 s (Table 1). Peak thrombin generation was elevated, indicative of decreased function of PS (Figure 1). In addition, peak thrombin level reverted to the normal range by adding increasing concentrations of PS (Figure 2), thereby confirming that the elevated generation of thrombin was a direct consequence of low PS activity. The ELISA showed that the study patient overall had a decreased level of free PS (Figure 3).
| aPPT Time | |
|---|---|
| Normal Patient Reference Range 30-40s | PS- deficient individual 28s |
Table 1: The Comparison between aPTT values (in seconds) of the normal range and the female PS deficient individual.
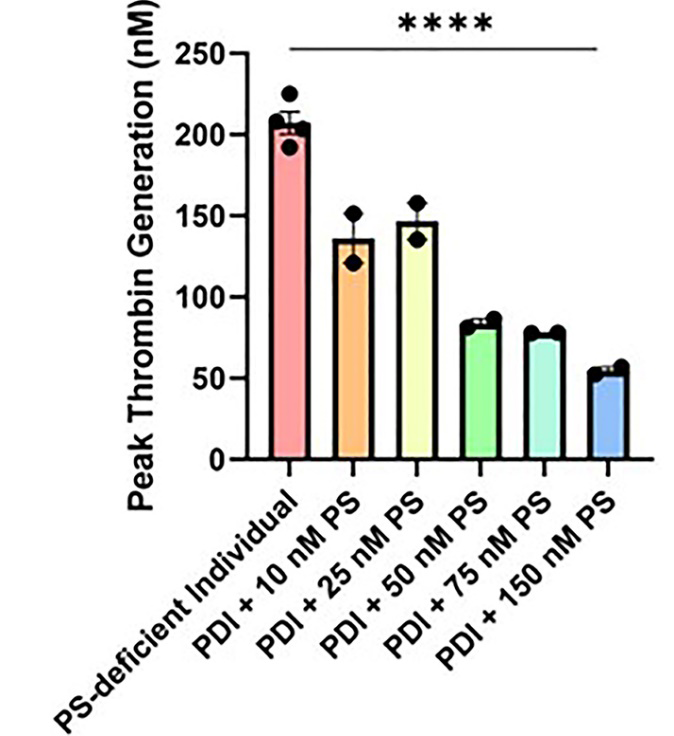
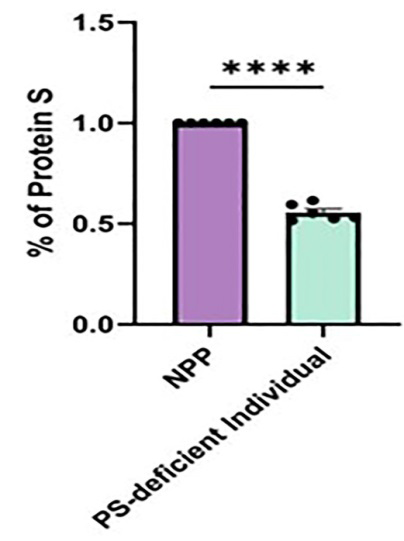
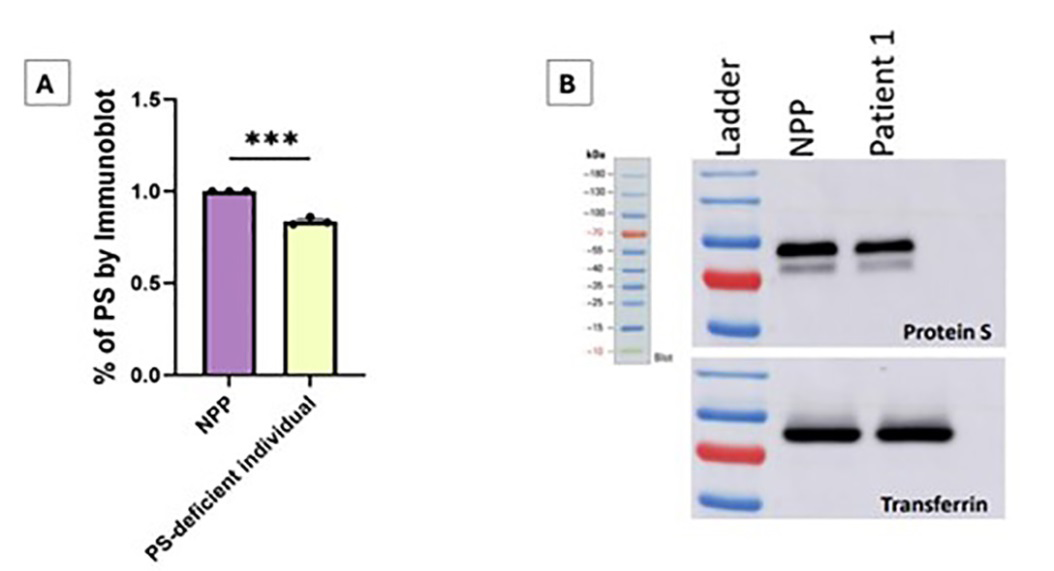
Figure 3: Part A is the percentage of PS in the patient compared to normal pooled plasma (NPP) with normal levels of PS as determined by immunoblot. Data was analyzed with an unpaired t test, with *** as p-value < 0.0001. Part B demonstrates immunoblot of total Protein S levels in normal pooled plasma (NPP) and the patient’s samples. Transferrin was used as a loading control with Sheep Anti-PS Antibody/Anti-sheep IgG.
Novel Genetic Mutations
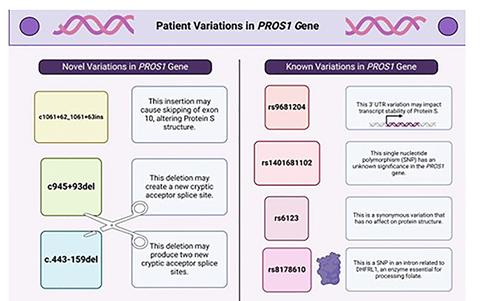
Sequencing of DNA isolated from the patient’s plasma and two controls revealed shared and novel mutations. Both the patient and controls had a single nucleotide polymorphism rs6123 on chromosome 3q11.1. This polymorphism is benign and has no effect on PS structure or function [28]. Other SNPs that similarly do not affect PS structure and function included variants in rs9681204, a SNP in the 3’-UTR of the PROS1 mRNA and SNP rs1401681102 in intron 5 (Figure 4).
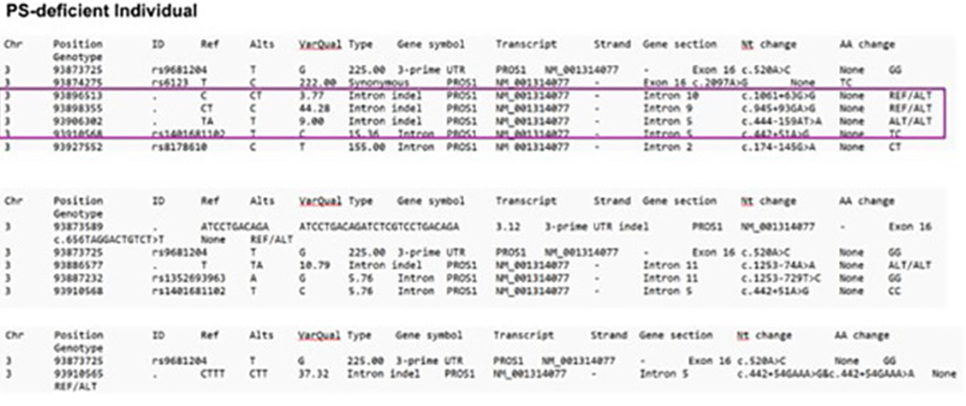
Figure 4: Patient variations compared to control 1 and control 2 individuals without PS deficiency is shown. Four differing variations in the patient compared to the control are outlined in purple. Each abbreviation is described as follows: Chr = chromosome, ID = identity, Ref = reference, Alt = alternates, VarQual = variation quality, Nt change = nucleotide change, AA change = amino acid change.
Novel variations in the PROS1 gene likely contributed to the woman’s PS deficiency and included a SNP mutation in rs8178610 in intron 2, which may also alter the structure of dihydrofolate reductase (DHFR). The patient had a deletion that may produce two new acceptor splice sites (intron 5 – c.444-157 and intron 5 – c.444-139). The c.443-159del variation may lead to splicing alterations according to the SSF, MaxEnt (+26.8%), and NNSPLICE (+6.0) predictors. Acceptor alteration may cause skipping of exon 6 or retention of part of intron 5. Another deletion was present as c.945+93del. This deletion may produce a new acceptor splice site and alter splicing, according to the NNSPLICE predictor. The variation may increase by 11.9% the possibility of retaining base pairs of intron 9 in the mRNA of splicing out all of exon 10. A novel intron insertion, c.1061+62_1061+63ins, may activate a donor site according to the three predictors: SSF (77,42), MaxEnt (5,89), and NNSPLICE (0,85). Donor site alteration may cause skipping of exon 10, thus producing partial intron retention. A summary of the variations is in Figure 5.
Discussion
Although exceedingly rare, hereditary PS deficiency is an autosomal dominant disorder with hundreds of recorded variants in the PROS1 gene. In this study, we identified three novel variants in the PROS1 gene on chromosome 3 from a woman who has a PS deficiency. These variations are present in cryptic splice sites, places in the genome which resemble physiological splice sites [28]. These sites normally indicate where introns are removed from DNA, but due to variations in the genetic sequence, new splice sites, also known as cryptic splice sites, arise and inappropriately impact pre- mRNA processing [28]. A deletion in the individual, c.444- 159del, may produce two new acceptor splice sites (intron 5 – c.444-157 and intron 5 – c.444-139). According to splice site predictions, potential for splicing alterations is elevated at 26%, preventing transcription of exon 6 or retention of base pairs in intron 5. Thus, a new isoform or nonfunctional version of PS would be produced.
Also, c.945+93del may produce a new acceptor splice site. Although splice site predictions indicate that this deletion may be less detrimental to PS pre-mRNA processing, this deletion may produce a new acceptor splice site and a potential alteration of splicing, according to the NNSPLICE predictor. The variation may increase by 11.9% the retention of base pairs of intron 9 by the mRNA or loss of exon 10 entirely. Likewise, these changes could result in an isoform or a nonfunctional protein. Such a variation would result in low production or malfunction of PROS1 protein products. A novel intron insertion, c.1061+62_1061+63ins, may introduce a donor splice site. A donor splice site is present in the upstream part of an intron (in direction 5’ to 3’) and c.1061+62_1061+63ins may lead to subsequent skipping of exon 10.
Conclusion
Given the function of PS as a cofactor for APC, TFPI, and a direct inhibitor of FIXa, PS prevents coagulation by a multi-pronged approach and is essential for appropriate hemostasis. Produced from hepatocytes, platelets, and endothelial cells, PS is synthesized and secreted at minute amounts, and the regulation of production and release is affected by several factors. Hormonal influences by estrogen and progesterone, and certain disease states affect level of PS. Furthermore, congenital PS deficiency can be categorized into three Types, Type I, Type II, and Type III, each with unique mechanisms leading to PS deficiency. Improving our understanding of the pathogenesis of inherited PS deficiency is essential for guiding future treatment for this life-threatening disease.
This study describes novel mutations in the PROS1 gene in an individual with Type I PS deficiency and the predicted impact of these mutations on PS pre-mRNA processing. Moving forward, further investigation of these mutations via experimentation is necessary to understand the true effect of the mutations on production of nonfunctional or under- expressed PS. Overall, discovery of these novel variants in the PROS1 gene not only advances our understanding of hereditary PS deficiency, but also the discovery expands opportunities for future gene therapy and treatment.
Studies in Humans
The work described has been conducted in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. All procedures were performed in compliance with relevant laws and institutional guidelines and have been approved by the appropriate institutional committees. Informed consent was obtained from the individual prior to data collection and experimentation.
Contribution
T.P. wrote the first draft, completed subsequent revisions, and compiled references; A.M. conducted most of the experiments; M.K., D.P.V, J.Z., and J.G. conducted experiments and genetic analysis; S.G. created figures; M.M. wrote the method and materials section; R.M. was responsible for final revisions and oversight of the project.
We thank Ma. Lorena Duhaylungsod, Vandana Sood, Rafika Yasmin, and Narender Kumar for their support and advice. This work was supported by 1R01HL151613-01A1 to RM.
Conflicts of Interest
The authors declare that no conflict of interest exists.
References
-
Scipio RG, Hermodson MA, Yates SG, Davie EW (1977) A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry 16(4): 698-706.
-
Comp P, Nixon R, Cooper M, Esmon C (1984) Familial protein S deficiency is associated with recurrent thrombosis. Journal of Clinical Investigations 74(6): 2082-2088.
-
Dahlback B (2018) Vitamin K-Dependent Protein S: Beyond the Protein C Pathway. Semin Thromb Hemost 44(2): 176-184.
-
Majumder R, Nguyen T (2021) Protein S: function, regulation, and clinical perspectives. Curr Opin Hematol 28(5): 339-344.
-
Saller F, Villoutreix BO, Amelot A, Kaabache T, Bonniec BF, et al. (2005) The gamma-carboxyglutamic acid domain of anticoagulant protein S is involved in activated protein C cofactor activity, independently of phospholipid binding. Blood 105(1): 122-130.
-
Melancon D, Nguyen T, Pereira V, Harman J, Chatterjee S, et al. (2024) Downregulation of protein S in patients with severe COVID-19 augments the prothrombotic state. Thromb Res 238: 129-131.
-
Werner LM, Criss AK (2023) Diverse Functions of C4b-Binding Protein in Health and Disease. J Immunol 211(10): 1443-1449.
-
Suleiman L, Negrier C, Boukerche H (2013) Protein S: A multifunctional anticoagulant vitamin K-dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer. Critical Reviews in Oncology Hematology 88(3): 637-654.
-
Dahlback B (2016) Pro and anticoagulant properties of factor V in pathogenesis of thrombosis and bleeding disorders. Int J Lab Hematol 38(1): 4-11.
-
Maurissen LF, Thomassen MC, Nicolaes GA, Tans G, Rosing J, et al. (2008) Re-evaluation of the role of the protein S-C4b binding protein complex in activated protein C-catalyzed factor Va-inactivation. Blood 111(6): 3034-3041.
-
Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP (2001) Structure and biology of tissue factor pathway inhibitor. Thromb Haemost 86(4): 959-972.
-
Mast AE (2016) Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arterioscler Thromb Vasc Biol 36(1): 9-14.
-
Plautz WE, Chattopadhyay R, Goldfeld EI, Simioni P, Arruda VR, et al. (2018) Padua FIXa resistance to Protein S and a potential therapy for hyperactive FIXa. Thromb Res 170: 133-141.
-
Schmidel DK, Tatro AV, Phelps LG, Tomczak JA, Long GL (1990) Organization of the Human Protein-S Genes. Biochemistry 29(34): 7845-7852.
-
Sankpal UT, Goodison S, Abdelrahim M, Basha R (2011) Targeting SP1 Transcription Factor in Prostate Cancer Therapy. Medicinal Chemistry 7(5): 518-525.
-
Hughes Q, Watson M, Cole V, Sayer M, Baker R, et al. (2007) Upregulation of protein S by progestins. J Thromb Haemost 5(11): 2243-2249.
-
Matthes B (1992) Acquired protein S deficiency. Clin Investig 70(6): 529-534.
-
Morange PE, Alessi MC, Barthet MC, Aillaud MF, Harle JR, et al. (1997) Acquired protein S deficiency, likely due to anti-PS autoantibodies, following a thrombotic event in a patient with a systemic lupus erythematosus. Thrombosis and Haemostasis 78(5): 1416-1417.
-
Kearon C, Crowther M, Hirsh J (2000) Management of patients with hereditary hypercoagulable disorders. Annu Rev Med 51: 169-185.
-
Pung P, Poort SR, Vos HL, Mahasandana C, Bertina RM, et al. (1998) Compound heterozygosity for one novel and one recurrent mutation in a Thai patient with severe protein S deficiency. Thromb Haemost 81(2): 189-192.
-
Mahasandana C, Suvatte V, Marlar RA, Johnson MJ, Jacobson LJ, et al. (1990) Neonatal purpura fulminans associated with homozygous protein S deficiency. Lancet 335(8680): 61-62.
-
Rosendaal FR (1999) Venous thrombosis: a multicausal disease. Lancet 353(9159): 1167-1173.
-
Gupta A, Tun AM, Gupta K, Tuma F (2024) Protein S Deficiency. StatPearls. Treasure Island (FL).
-
Campello E, Spiezia L, Simion C, Tormene D, Camporese G, et al. (2020) Direct Oral Anticoagulants in Patients with Inherited Thrombophilia and Venous Thromboembolism: A Prospective Cohort Study. J Am Heart Assoc 9(23): e018917.
-
Zhou J, Shen W, Gu Y, Li M, Shen W (2020) Compound heterozygous mutations identified in severe type I protein S deficiency impaired the secretion of protein S. J Clin Pathol 73(1): 7-13.
-
Agarwal SA, Santhanam J, Degapudi S (2022) A Case of Type 2 Protein S Deficiency Presenting as Cerebral Venous Thrombosis (CVT) in an 18-Year-Old Female. Cureus 14(8): e28221.
-
Kimura R, Honda S, Kawasaki T, Madoiwa S, Sakata Y, et al. (2006) Protein S-K196E mutation as a genetic risk factor for deep vein thrombosis in Japanese patients. Blood 107(4): 1737-1738.
-
Baten AK, Chang BC, Halgamuge SK, Li J (2006) Splice site identification using probabilistic parameters and SVM classification. BMC Bioinformatics 5(5): S15.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies