The Chloroplast Genome Sequence Characteristics Analysis of Berberis diaphana Maxim
Berberis diaphana Maxim (B. diaphana), an important medicinal deciduous shrub, is narrowly endemic to China. In the present study, the complete chloroplast (cp) genome of B. diaphana was sequenced and analyzed. The complete cp genome of B. diaphana is 166, 225 bp in length, presenting a quadripartite structure comprising a large single copy region (LSC 73, 596 bp), a small single copy region (SSC 18, 656 bp), and a pair of inverted repeats regions (IRa/ IRb: 36, 954 bp each). The genomewide GC content was 38.07%, LSC made up 32.59%, SSC made up 32.59%, and IR made up 41.02%. The genome encodes 144 genes, including 99 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. A total of 98 simple sequence repeats (SSRs) was identified. Codon usage analysis showed that the genome tended to use codon ending in A/U. Phylogenetic relationships of 20 species inferred that B. diaphana is sister to Berberis weiningensis. The genome comparison revealed that there is a wide variability of the junction sites, and there is higher divergence in the SSC and IRa/ IRb regions than in LSC regions. This study identified the unique characteristics of the B. diaphana cp genome, which will provide valuable information for further studies as well as molecular identification approaches for this important medicinal plant.
Introduction
Chloroplasts are important organelles of green plants that are responsible for photosynthesis [1]. The chloroplast genome has independent nuclear genome, and has the characteristics of haploid inheritance, relatively small genome, slow mutation rate, and sufficient plant polymorphism, which makes it conducive to the study of plant phylogeny. CP genome-based molecular barcoding enables accurate identification of plants at the species and population level [2]. In recent years, with the rapid development of high-throughput sequencing technology, the assembly of chloroplast genomes has become convenient and cheap, and phylogenetic reconstruction based on chloroplast genomes has led to a better understanding of the evolutionary relationship of angiosperms.
Berberis Linn. constitutes the largest genus in the family Berberidaceae. The plants of this genus are distributed widely across India, Pakistan, China, Japan, Europe, and South America [3]. The Berberis L. genus comprises approximately 500 species of evergreen or deciduous simple-leaved shrubs [4]. The Qinghai Province of China is considered an important distribution region of Berberis plants as it is habitat to 13 species and one variant of this genus [5]. The stem, leaf, flower, and fruit morphologies of Berberis species demonstrate high diversity due to the diversity of species and distribution area [3]. Barberry plants have fruits, flowers, leaves, stems and roots. they are often used for medicinal purposes because of their various bioactive ingredients, such as flavonoids (quercetin, myricetin, and luteolin) Mustafa K, et al. [6], Nazir N, et al. [7], phenolics (chlorogenic, caffeic, and vanillic acids) Çakır Ö, et al. [8], Dong ZY, et al. [9], anthocyanin Sharif A, et al. [10], Arena ME, et al. [11], alkaloids (berberine and palmatine) Mustafa K, et al. [6], Bisht A, et al. [12], tannins, carbohydrates, terpenoids, and steroids [13, 14, 15].
One of these species is B. diaphana, which is distributed mainly in the Qinghai, Shaanxi and Gansu at an altitude of 1620-3600 m. B. diaphana has been used for the treatment of diarrhea, frequent urination, diabetes, trachoma, and nephritis for centuries in traditional Tibetan medicinal system [16]. Berberine derived from the roots and stems of B. diaphana is well recognized for its effective antineoplastic activities and therapeutic effect in type 2 diabetes [17, 18]. Recently, a few studies have been conducted to reveal the phylogenetic relationships and the evolutionary history of B. diaphana [19, 20]. Nonetheless, the possible relationship among the different species of Berberis genus remains uncertain so far. In this context, investigations on the cp genome would provide valuable information on the botanic taxonomy of Berberidaceae, and would also contribute to determining the evolutionary position and protecting the germplasm resources of B. diaphana.
Material and Methods
Plant Material
In the present study, fresh leaves of B. diaphana were collected from Xunhua County (102.6749°E, 35.7917°N, Qinghai, China) and stored immediately at liquid nitrogen. A specimen was deposited at the Tibetan medicine research center at Qinghai University under the voucher number TMSGS21003.
DNA Extraction, Sequencing, and Cp Genome Assembly
DNA was extracted from the collected B. diaphana leaves using the modified CTAB method [21]. The high-quality DNA obtained was used for library construction and sequencing in Beijing Biomarker Technologies Co. Ltd. using Illumina Hiseq X Ten, which was followed by de novo assembling of the genome using SPAdes [22].
Annotation of the B. Diaphana Cp Genome
B. diaphana cp genome annotation was performed via the Cp GAVAS pipeline [23]. The assembled complete cp genome of B. diaphana was deposited to NCBI under accession number MZ962404. The circular gene map and cis- and trans- splicing genes were visualized in CPGview in the B. diaphana cp genome [24]. The GC content was showed using online tools proksee (https://proksee.ca/). Relative synonymous codon usage (RSCU) was determined by CodonW version 1.4.4. The online software Reputer Kurtz S, et al. [25] was used to identify the repeat sequences (n ≥ 30 bp). The repeat matches were categorized as follows: forward (direct) match (F), reverse match (R), complement match (C) and palindromic match (P). SSRs in the cp genome were identified using the online microsatellite identification tool MISA (https://webblast.ipk-gatersleben. de/misa/). The parameters were set to ten repeat units for mononucleotide SSRs, five repeat units for dinucleotide SSRs, four repeat units for trinucleotide SSRs, and three repeat units each for tetranucleotides, pentanucleotide and hexanucleotides SSRs.
Phylogenetic Analysis
To determine the phylogenetic relationships among Berberidaceae species, we downloaded 19 cp genome sequences from the NCBI database for phylogenetic analysis. The phylogenetic tree was constructed using the maximum- likelihood (ML) method in the Mega7 program using 1000 bootstrap [27].
Results
Genome Features of B. diaphana
The phenotype of B. diaphana is shown in Figure 1. Filtering of the raw sequencing data yielded a total of 11, 791, 180 clean paired-end reads in B. diaphana cp genome. There were 3.5 G bases, of which 93.05% of bases had a quality score higher than Q30, and 97.44% of bases had a quality score higher than Q20.

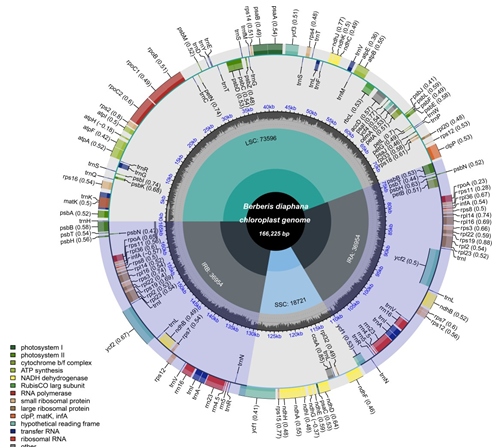
Figure 2: Chloroplast genome overall features map of B. diaphana. From the center outward: the first track shows the small single-copy (SSC), inverted repeat (IRa and IRb), and large single-copy (LSC) regions. The GC content along the genome is plotted on the second track. The genes are shown on the third track. The optional codon usage bias is displayed in the parenthesis after the gene name. Genes are color-coded by their functional classification. The transcription directions for the inner and outer genes are clockwise and anticlockwise, respectively. The functional classification of the genes is shown in the bottom left corner.
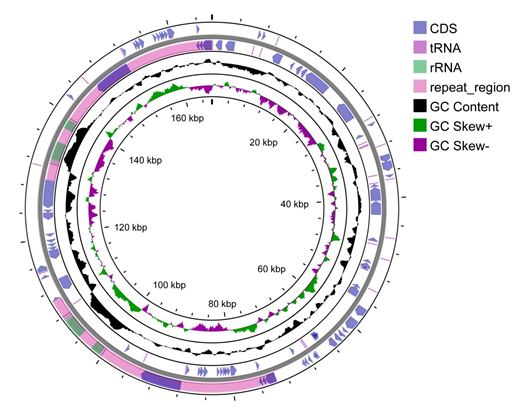
Similar to the other angiosperm that have been sequenced, B. diaphana also had a cp genome with a typical quadripartite circular structure. The genome was 166, 225 bp in length, which included 73, 596 bp of a large single- copy region (LSC), 18, 656 bp of a small single-copy region (SSC), and two inverted repeat sequence regions (IRa/ IRb: 36, 954 bp each) (Figure 2). The overall GC content is
38.07%. The IR regions have a relatively higher GC content (41.02%) compared with LSC and SSC (32.59%) regions (Figure 3). The cp genome contained a total of 144 genes, including 99 protein-coding genes, 37 tRNA genes, and 8 rRNA genes (Table 1). Among them 31 genes are duplicated in IRs, including 6 tRNA genes, 4 rRNA genes, and 21 protein- coding genes (Table 1).
| Category for genes | Group of genes | Name of genes | Number of genes |
|---|---|---|---|
| Transcription and translation related genes | Ribosomal RNA genes | trnY-GUA, trnW-CCA, trnV-UAC, trnV-GAC×2, trnT-UGU, trnT- GGU, trnS-UGA, trnS-GGA, trnS-GCU, trnR-UCU, trnR-ACG×2, trnQ-UUG, trnP-UGG, trnN-GUU×2, trnM-CAU, trnL-UAG, trnL-UAA, trnL-CAA, trnL-CAA, trnK-UUU, trnI-GAU×2, trnI- CAU×2, trnH-GUG, trnG-UCC, trnG-GCC, trnfM-CAU, trnF-GAA, trnE-UUC, trnD-GUC, trnC-GCA, trnA-UGC×2 | 37 |
| Photosynthesis- | Transfer RNA genes | rrn23×2, rrn16×2, rrn5×2, rrn4.5×2 | 8 |
| Small subunit of ribosome | rps2, rps3×2, rps4, rps7×2, rps8×2, rps11×2, rps12×2, rps14, rps15, rps16, rps18, rps19×2 | 18 | |
| Large subunit of ribosome | rpl2×2, rpl14×2, rpl16×2, rpl20, rpl22×2, rpl23×2, rpl32, rpl33, rpl36×2 | 15 | |
| DNA dependent RNA polymerase | rpoA×2, rpoB, rpoC1, rpoC2 | 5 | |
| Subunits of photosystem І | psaA, psaB, psaC, psaI, psaJ | 5 | |
| Subunits of photosystem ІІ | psbA, psbB×2, psbC, psbD, psbE, psbF, psbH×2, psbI, psbJ, psbK, psbL, psbM, psbN×2, psbT×2, psbZ, ycf3 | 19 | |
| Subunits of cytochrome | petA, petB, petG, petL, petN | 5 | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | 6 |
Table 1: Genes with introns in the _B. diaphana_ cp genome.
ATP-dependent protease subunits P gene ClpP 1
Large subunits of RuBisCO rbcL 1
Subunits of NADH dehydrogenase ndhA, ndhB×2, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK 12 Maturase matK 1 Envelope membrane protein cemA 1 Other genes Subunits of acetyl-CoA- carboxylase accD 1 C-type cytochrome synthesis gene ccsA 1 Translation initiation factor 1 infA×2 2 Unknown function Conserved open reading frames ycf4, ycf2×2, ycf1×2 6
- ×2 two gene copies in the IRs
Table 2: Annotated genes in the B. diaphana cp genome.
| Gene | ExonI(bp) | IntronI(bp) | ExonII(bp) | IntronII(bp) | ExonIII(bp) |
|---|---|---|---|---|---|
| trnK-UUU | 37 | 2505 | 35 | ||
| rps16 | 40 | 905 | 197 | ||
| trnG-UCC | 23 | 723 | 48 | ||
| atpF | 145 | 725 | 410 | ||
| rpoC1 | 453 | 760 | 1611 | ||
| ycf3 | 126 | 726 | 228 | 747 | 153 |
| trnL-UAA | 35 | 488 | 50 | ||
| trnV-UAC | 39 | 590 | 35 | ||
| clpP | 98 | 732 | 294 | 732 | 241 |
| petB | 6 | 725 | 6 | ||
| rpl16* | 9 | 830 | 399 | ||
| rpl2* | 400 | 659 | 434 | ||
| ndhB* | 775 | 691 | 761 | ||
| ndhA | 556 | 1048 | 539 | ||
| trnA-UGC* | 38 | 800 | 35 | ||
| trnI-GAU* | 37 | 944 | 35 | ||
| rps12* | 114 | 27575 | 232 | 794 | 26 |
Table 3: Genes with introns in the _B. diaphana_ cp genome.
* two gene copies in the IRs Table 2: Genes with introns in the B. diaphana cp genome.

Figure 4: The cis- and trans- splicing genes in the B. diaphana cp genome. A. The cis-splicing genes in the B. diaphana cp genome. The gene names are shown on the left, and the gene structures are on the right. The exons are shown in black; the introns are shown in white. The arrow indicates the sense direction of the gene. B. The trans- splicing gene rps12 in the B. diaphana cp genome. Two of them are duplicated as they are located in the IR regions.
In the B. diaphana cp genome, 23 intron-containing genes were annotated, including 8 tRNA genes and 15 protein-coding genes (Table 2). All intron-containing genes include 6 duplicated genes in the IR regions, with 2 tRNA genes (trnA-UGC and trnI-GAU) and 4 protein-coding genes (rpl16, rpl2, ndhB, rps12). Except for ycf3, clpP, and rps12 (duplicated) with 2 introns and the other 19 genes with 1 intron. The longest intron length is trnK-UUU (2505 bp), and the shortest intron length is trnL-UAA (499 bp) (Table 2). Furthermore, 13 cis-splicing genes were predicted in the B. diaphana cp genome, the structure of these genes were showed in Figure 4A. The rps12 gene is a trans-splicing gene with the 5’ end in the LSC region and the 3’ end in the IR region (Figure 4B).
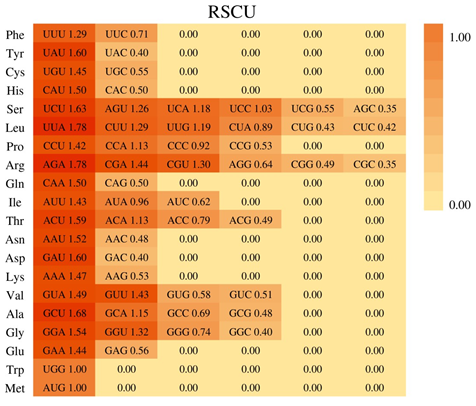
The relative frequency of synonymous codons of the B. diaphana cp coding sequence was estimated. The results shows that all genes are encoded by 28, 308 codons, and the 4 most frequently used codons were AAA (lysine), AUU (isoleucine), GAA (glutamic acid), and UUU (Phe), pertaining to 1, 165 (4.40%), 1, 142 (4.31%), 1, 097 (4.14%), and 1, 027 (3.88%) codons, respectively (Table 1 & Figure 4). The most frequently used amino acids were leucine (2, 888), whereas cysteine was the least abundant, with only 325 hits. Codons ending in A and U are very common. Except for rnL-CAA and trnS-GGA, all codons (RSCU > 1) end in A or U. And A- and U-ending codons accounted for 69.06% among all codons (Table 1).
Long Repeats and Simple Sequence Repeats (SSRS) Analysis
A total of 151 (≥30 bp) long repeats were identified in the cp genome of B. diaphana, including 88 forward repeat, 46 palindromic repeat and 17 reverse repeats, no complementary repeats are present (Table 2). Most repeats ranged from 30 to 73 bp in length (94.03%). Then we studied the type and number of SSR in the B. diaphana cp genome (Table 3). A total of 98 SSRs were found in B. diaphana, including 70 mononucleotide SSRs (71.43%), 10 dinucleotide SSRs (10.20%), 3 trinucleotide, 7 tetranucleotides, and 8 hexanucleotides repeats. The mononucleotide A and T repeat units accounted for the largest portion, indicating that the SSR sequence of B. diaphana cp genome prefers A/T base.
| Repeat types | SSR Motif | Length/bp | Number | Total |
|---|---|---|---|---|
| Mononucleotide -71.43% | A | 9/10/11/12/13/15/16 | 9/6/4/4/3/1/3 | 30 |
| T | 9/10/12/13/15/16/17/25 | 15/7/5/6/1/3/1/1 | 40 | |
| AT | 4/5 | 2/1 | 3 | |
| Dinucleotide -10.2% | CT | 4 | 1 | 1 |
| TA | 4 | 5 | 5 | |
| TG | 5 | 1 | 1 | |
| Trinucleotide -3.06% | CTT | 9 | 1 | 1 |
| TAT | 3/4 | 1/1 | 2 | |
| AAAT | 3 | 1 | 1 | |
| Tetranucleotides -7.14% | AATA | 3 | 1 | 1 |
| ATTA | 3 | 1 | 1 | |
| CCAT | 3 | 1 | 1 | |
| TTAA | 3 | 2 | 2 | |
| TTTA | 3 | 1 | 1 | |
| AGTGAT | 3 | 1 | 1 | |
| ATAGAA | 3 | 1 | 1 | |
| ATTTTT | 3 | 1 | 1 | |
| Hexanucleotides (8.16%) | CTCGTC | 3 | 1 | 1 |
| TATCAC | 3 | 1 | 1 | |
| TCAATA | 3 | 1 | 1 | |
| TTGATA | 3 | 1 | 1 | |
| TTTCTA | 3 | 1 | 1 |
Table3: Types and number of SSR of B. diaphana cp genome.
Comparative Analysis of Genomic Structure
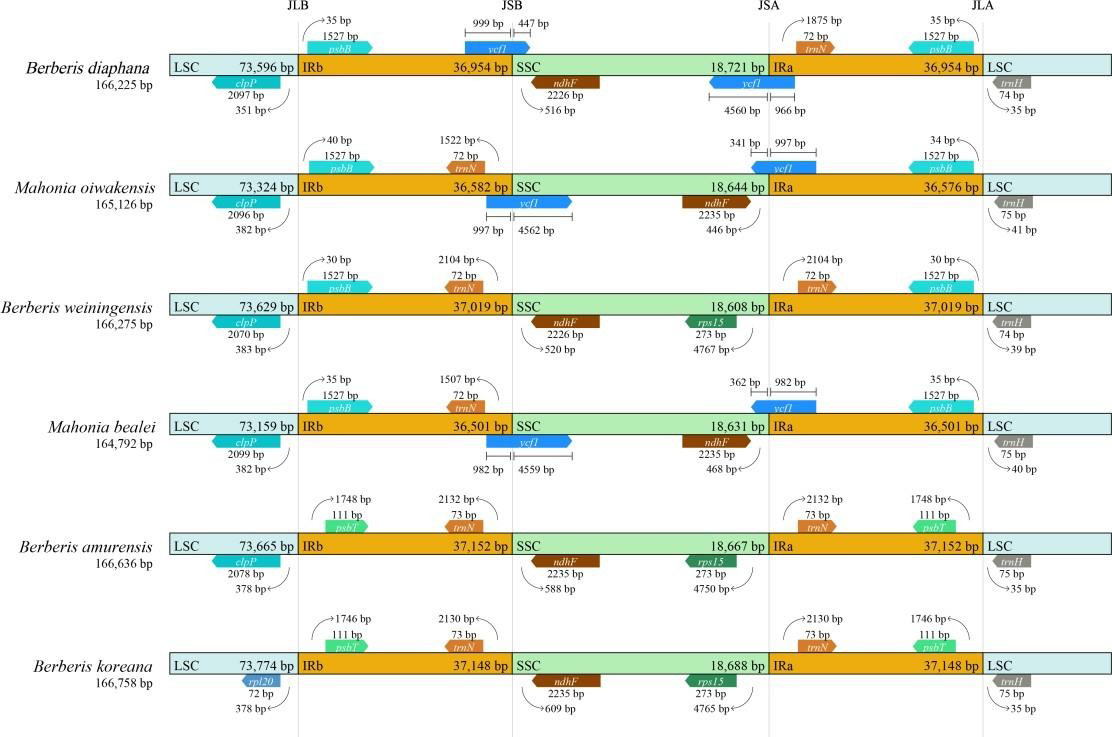
To further resolve the structural evolutionary history of the cp genomes of the Berberidaceae, we compared the IR/ SSC and IR/LSC junctions across six selected Berberidaceae species, including Mahonia oiwakensis, Berberis weiningensis, Mahonia bealei, Berberis koreana, Berberis amurensis and B. diaphana. In this case, JLB symbolizes the joint LSC/IRb, while JSB symbolizes the joint SSC/IRb, JSA represents the joint SSC/IRa, and JLA symbolizes the joint LSC/IRa. The results showed that these 6 species had similar genome sequence lengths, and there were certain differences in the location and type of boundary genes (Figure 6). We observed that the B. diaphana exhibited same JLB and JLA junction sites compared with Mahonia bealei and Mahonia oiwakensis, and the genes on both sides of the boundary are the same.
Both JSB and JSA junction sites were within the ycf1 gene, but the degree of gene expansion varies. At the JSB boundary, the ycf1 gene was located at 999bp in the IRb region and 447bp in the SSC region in B. diaphana, while at the JSA boundary, the ycf1 gene was located at 966bp in the IRa region and 4560bp in the SSC region. Compared with the other 5 species of Berberidaceae, B. diaphana was missing the trn N gene in the IRb region.
Phylogenetic Analysis
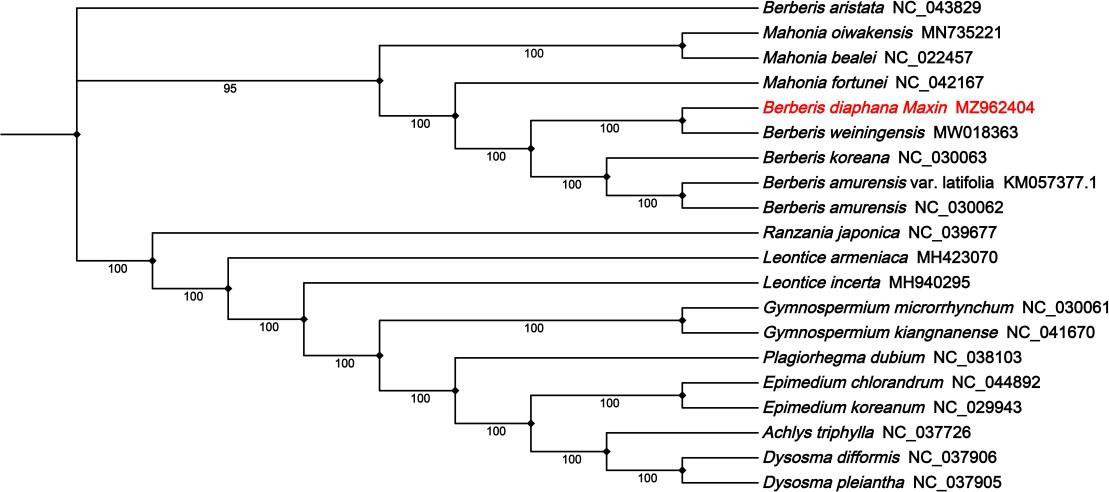
The phylogenetic analysis of the complete cp genome sequences of B. diaphana and 19 other related species within the Berberidaceae species was conducted (Figure 7). The evolutionary tree was separated into three clusters. The phylogenetic tree showed that berberis aristata were clustered on a single branch. We found that B. diaphana formed an independent branch with Mahonia oiwakensis, Mahonia bealei, Mahonia fortune, Berberis weiningensis, Berberis amurensis var. latifolia, Berberis amurensis and Berberis koreana. And B. diaphana was closely related to Berberis weiningensis under 100% bootstrap support values. Meanwhile, Ranzania japonica, Leontice incerta, Leontice armeniaca, Gymnospermium microrrhynchum, Gymnospermium kiangnanense, Plagiorhegma dubium, Epimedium chlorandrum, Epimedium koreanum, Achlys triphylla, Dysosma difformis and Dysosma pleiantha were clustered on a branch.
Discussion
In this study, we assembled, annotated and analyzed the complete cp sequence of B. diaphana. Then we analyzed its features, GC content, gene structure, and SSRs. The complete cp genome of B. diaphana displays a typical quadripartite structure with a total length of 166, 225 bp, including one SSC, one LSC, and two IRs (Figure 2). In total, 144 genes, including 99 protein-coding genes, 37 tRNA genes, and 8 rRNA genes (Table 1), were identified in our research. The overall GC content is similar to that observed for other Berberidaceae species, both around 38% [20, 28]. It has been shown that GC skewness correlates with DNA lead chains, lag chains, replication origins, and replication terminals, which is an outstanding indicator of species affinity [29]. In our research, it is obvious that the GC content of the IR region is higher than that of other regions (LSC, SSC) (Figure 3); this phenomenon is very common in other flowering plants [30, 31]. The 5′-end exon of rps12 gene is located in the LSC region, while the 3′-end exon is located in the IR region in B. diaphana (Figure 4B), this result is similar to that of the congeneric specie [30, 32].
The codon usage bias of cp genomes may be a result of selection and mutation [33]. The frequency of codon usage was estimated for the B. diaphana cp genome in this study. We found that all genes are encoded by 28, 308 codons, and the 4 most frequently used codons were AAA (lysine), AUU (isoleucine), GAA (glutamic acid), and UUU (Phe) (Figure 4); among these codons, A- and U- ending codons accounted for 69.06% among all codons (Table 1). The results are similar to those reported in other angiosperms Jian HY, et al. [34], Liu L, et al. [35], Li Y, et al. [36], and it is possible to use these features of codon usage preference in order to better understand exogenous gene expression and evolution of the cp genome.
SSRs are inevitable and highly variable products of genome replication, playing an important role in genome evolution and recombination Subramanian S, et al. [37], and can participate in gene expression, regulation, and component functions such as transcriptional activation [38]. CP- specific SSR has the characteristics of non-recombinant, haploid, monoparental inheritance and low substitution rate, which is commonly used in population genetics, species identification, and evolutionary process research. This study found mononucleotide SSRs were the richest (occupied 71.43%), and the mononucleotide A and T repeat units occupied the highest portion, these results are consistent with a previous study and verify the hypothesis that cpSSRs are generally composed of short polyadenine (polyA) or polythymine (polyT) repeats and rarely contain tandem guanine (G) or cytosine (C) repeats [39, 40].
The expansion and contraction of cp genome is a common evolutionary phenomenon in plants [41]. We found that there were differences in the size and boundary genes of the LSC, SSC, and IR regions among six Berberidaceae species, and there is higher divergence in the SSC and IRa/ IRb regions than in LSC regions. It was speculated that this phenomenon may be caused by the distribution area and long-term evolutionary process of the species. The phylogenetic positions of 20 cp genomes were successfully analyzed with the support of full bootstrap at almost all nodes. A phylogenetic tree was constructed for the data by ML. The results show that B. diaphana has the closest relationship with Berberis weiningensis.
Conclusion
We analyzed and illustrated the complete cp genome of B. diaphana. The cp genome is conservative and similar to other species of Berberis. These results provide a reference for the complete assembly of the cp genome of Berberidaceae, which would be useful to subsequent studies that conducted on conservation genetics, phylogeny and molecular breeding in relation to genus Berberis.
References
-
Brunkard JO, Runkel AM, Zambryski PC (2015) Chloroplasts extend stromules independently and in response to internal redox signals. Proceedings of the National Academy of Sciences of the United States of America 112(32): 10044-10049.
-
Hernandez-Leon S, Gernandt DS, Rosa JAPdl, Jardon- Barbolla L (2013) Phylogenetic relationships and species delimitation in pinus section trifoliae inferrred from plastid DNA. PLoS One 8: e70501.
-
Ahrendt L (1961) Berberis and Mahonia, a taxonomic revision. Botanical Journal of the Linnean Society 57(369): 1-410.
-
Ying JS (2011) Editorial Board for Flora of China. Flora of China (revised English version). In: Ying JS (Ed.), Beijing: Science Press 19: 715-771.
-
Li YJ (1987) The woody palnts of Qinghai (Version 1). Qinghai: People’s Publishing House 201.
-
Mustafa K, Mohamed H, Shah AM, Yu S, Akhlaq M, et al. (2020) In vitro anticancer potential of Berberis lycium Royle extracts against human hepatocarcinoma (HepG2) cells. Biomed Research International 2020: 8256809.
-
Nazir N, Rahman A, Uddin F, Khan Khalil AA, Zahoor M, et al. (2021) Quantitative Ethnomedicinal Status and Phytochemical Analysis of Berberis lyceum Royle. Agronomy 11(1): 130-142.
-
Çakır Ö, Karabulut A (2020) Comparison of two wild-grown Berberis varieties based on biochemical characterization. Journal of Food Processing and Preservation 44(11): 14844-14854.
-
Dong ZY, Zeng QH, Wei L, Guo X, Sun Y, et al. (2022) Berberisides A-D: three novel prenylated benzoic acid derivatives and a clerodane glycoside from Berberis tsarica aherndt. Natural Product Research 36(8): 1996- 2001.
-
Sharif A, Niakousari M, Mortazavi SA, Elhamirad AH (2019) High-pressure CO2 extraction of bioactive compounds of barberry fruit (Berberis vulgaris): process optimization and compounds characterization. Journal of Food Measurement and Characterization 13: 1139-1146.
-
Arena ME, Giordani E, Bustamante G, Radice S (2021) Variability in fruit traits and anthocyanin content among and within populations of underutilized Patagonian species Berberis microphylla G. Forst. Journal of Berry Research 11(1): 35-50.
-
Bisht A, Giri L, Belwal T, Pandey A, Bahukhandi A, et al. (2021) In vitro propagation and antioxidant potential of Berberis asiatica from Western Himalaya. An International Journal Dealing with all Aspects of Plant Biology 156(2): 490-496.
-
Hostalkova A, Marikova J, Opletal L, Korabecny J, Hulcova D, et al. (2019) Isoquinoline alkaloids from Berberis vulgaris as potential lead compounds for the treatment of Alzheimer’s disease. Journal of Natural Products 82(2): 239-248.
-
Rahim BZ (2019) Nutritional and phytochemical screening of wild fruit of _Berberis baluchistanica_-an endemic species To Pakistan. Applied Ecology and Environmental Research 17(6): 12697-12707.
-
Landry K B, Azam S, Rehman S, Tariq S, Iqbal B, et al. (2021) Phytochemical analysis of _Berberis lyceum_ methanolic extract and its antiviral activity through the restoration of MAPK signaling pathway modulated by HCV NS5A. Asian Pacific Journal of Tropical Biomedicine 11(3): 132-140.
-
Qinghai, Sichuan, Gansu, Yunnan, Xinjiang (1979) Tibetan Medicine Standards. Health Bureau of Tibet, Qinghai People’s Publishing Press, Xining 11.
-
Zhang YF, Li XY, Zou DJ, Liu W, Yang JL, et al. (2008) Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. Journal of Clinical Endocrinology & Metabolism 93(7): 2559-2565.
-
Elmerahbi R, Liu YN, Eid A, Daoud G, Hosry L, et al. (2014) _Berberis libanotica_ Ehrenb extract shows anti-neoplastic effects on prostate cancer stem/progenitor cells. PLoS One 9(11): e112453.
-
He PZ, Ma Q, Dong MF, Yang ZP, Liu LX (2019) The complete chloroplast genome of _Leontice incerta_ and phylogeny of Berberidaceae. Mitochondrial DNA Part B-Resources 4(1): 101-102.
-
Feng T, Xiao QY, Li WJ, Zhang C, He Y, et al. (2021) The complete chloroplast genome of _Berberis weiningensis_ (Berberidaceae), an endemic and traditional Chinese medicinal herb. Mitochondrial DNA Part B 6(3): 1175- 1177.
-
Arseneau JR, Steeves R, Laflamme M (2016) Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Molecular Ecology Resources 17(4): 686-693.
-
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19(5): 455-477.
-
Liu C, Shi L, Zhu Y, Chen H, Zhang J, et al. (2012) CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13: 715.
-
Liu S, Ni Y, Li J, Zhang X, Yang H, et al. (2023) CPGView: A package for visualizing detailed chloroplast genome structures. Molecular Ecology Resources 23(3): 694- 704.
-
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, et al. (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research 29(22): 4633-4642.
-
Amiryousefifi A, Hyvönen J, Poczai P (2018) IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34(17): 3030- 3031.
-
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution 33(7): 1870- 1874.
-
Xiao QY, Feng T, Yu Y (2020) The complete chloroplast genome of _Mahonia oiwakensis_ (Berberidaceae), a traditional Chinese medicinal plant. Mitochondrial DNA Part B 5(1): 692-694.
-
Necsulea A, Lobry JR (2007) A new method for assessing the effect of replication on DNA base composition asymmetry. Molecular Biology and Evolution 24(10): 2169-2179.
-
Shen J, Li X, Chen X, Huang X, Jin S (2022) The complete chloroplast genome of Carya cathayensis and phylogenetic analysis. Genes 13(2): 369.
-
Liu HY, Yu Y, Deng QY, Li J, Huang ZX (2018) The chloroplast genome of _Lilium henrici_: Genome structure and comparative analysis. Molecules 23(6): 1276.
-
Hu Y, Chen X, Feng X, Woeste KE, Zhao P (2016) Characterization of the complete chloroplast genome of the endangered species _Carya sinensis (Juglandaceae)_. Conservation Genetics Resources 8(4): 467-470.
-
Morton BR (2003) The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. Journal of Molecular Evolution 56: 616-629.
-
Jian HY, Zhang YH, Yan HJ, Qiu XQ, Wang QG, et al. (2018) The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species. Molecules 23(2): 389.
-
Liu L, Wang Y, He P, Pan Li, Lee J, et al. (2018) Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 19: 235.
-
Li Y, Sylvester SP, Li M, Zhang C, Li X, Duan Y, Wang X, et al. (2019) The complete plastid genome of _Magnolia_ _zenii_ and genetic comparison to Magnoliaceae species. Molecules 24(2): 261.
-
Subramanian S, Mishra RK, Singh L (2003) Genome- wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biology 4: R13.
-
Legendre M, Pochet N, Pak T, Verstrepen KJ (2007) Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Research 17(12): 1787-1796.
-
Shen X, Wu M, Liao B, Liu Z, Bai R, et al. (2017) Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant _Artemisia annua._ Molecules 22(8): 1330.
-
Wang L, Wuyun TN, Du H, Wang D, Cao D (2016) Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genetics & Genomes 12: 12.
-
Plunkett GM, Downie SR (2000) Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Systematic Botany 25(4): 648-667.
- Enhancement of Vegetative Growth and Fruit Yield in Cucumber (Cucumis sativus L.) via Spiritual Blessing (Biofield) Energy Intervention
- Production of Açaí (Euterpe oleracea Mart.) under Different Agroforestry System Management Intensities in Amazonian Floodplain (Varzea) Forests
- Coffee and the Production Region: What is the Secret to the Expression "Quality"?
- Experiential Agripreneurship Training in Sub-Saharan Africa: Integrating a Business Incubator into Postgraduate Livestock Education at the University of Buea
- Advances in Agricultural High-Quality Development
- Linking Compost Residue to ABAGE in Plants - a Short Note