Computerized Systems Validation (CSV) in Biopharmaceutical Industries
The biopharmaceutical industries has more and more used computers to support and accelrate producing of their products. Computer systems also are accustomed support routine offer of high quality products to boost production process performance, scale back production prices, and improve product quality. it's vital that these systems square measure suitable purpose from a business and restrictive perspective. Regulatory authorities treat a lack of regulatory computer system compliance as a serious GxP deviation. The objective of regulated computer systems includes systems used to manage data or support descion making subject to review by regulated authorities whether they are being submitted because its impact on quality or on business. Investments in computer systems supporting the quality controls to ensure that the process is followed correctly, reducing human error and the need to conduct manual checks, Standardization of practices to build consistent ways of working, Speed-up of process cycle times by reducing wait times and by improved scheduling...etc.Computer systems shouldn't be enforced only for restrictive compliance; operational advantages must always be exploredas well. “U.S. Code of Federal Regulation 21 CFR Part 600, 606, and 610†and “EU Directive 2003/94/EEC†are the prominent regulations reqested CSV, while “Volume 4 Good Manufacturing Practice Medicinal Products for Human and Veterinary Use - Annex 11: Computerised Systems†considered the main guidlines for CSV in biopharmaceutical industries in European Union. This paper aims to provide simplifed guidance on the basic requireents for computer system validation (CSV) based on the latest regulatory developments and industry trends. In conclusion, CSV has the great impact on the processes improvement. Also the critical parameters of computer systems validation for biopharmaceutical indsutries are highlighted.
Introduction
In 1990, when two European pharmaceutical manufacturers failed to meet computer compliance expectations and were momentarily forbidden from exporting their products to the United States, the problem of computer system validation assumed a high profile in Europe, EU requirements for computer systems compliance were issued a few years later in 1993 and can be found in Annex 11 in the EU GMPs [1]. So that biopharmaceutical companies should appoint a senior management representative with particular responsibility for ensuring the implementation of computer compliance requirements. This person, often graded as a Director, is a must-hearted champion of GxP’s cause. This senior position’s power and accountability should be obviously described and recorded. It is anticipated that the senior manager responsible for regulatory compliance will hire skilled and experienced personnel and guarantee that the CSV requirements carried out is carried out correctly and efficiently [2]. The failure to comply with regulations can has significant financial implications. Noncompliance issues may lead to delays in the issue of a license or its withdrawal and thus an embargo on the distribution of companies products in the relevant marketplace [3]. Successful validation relies on a number of fundamental supporting procedures being operated satisfactorily. These include instruction, document management, change control, configuration management, traceability specifications, self-inspection, and deviation management. Computer Systems Validation (CSV) is central to the life sciences industry [4].
Importance of CSV
Apart from being a regulatory requirement as set out by various regulatory authorities and practices such as the FDA, EMA, GCP, GLP, GMP and all the Predicate Rules; CSV is also very important to implement because not doing so will result in costly consequences such as
- Having a 483 form issued.
- Getting warning letter from FDA.
More than anything else, implementation of CSV is also important because it ensures that the data is accurate and the information, secure. Implementing Computer Systems Validation is also an important step in making sure that the organization restricts or prevents any loss of revenue from its main activities or from the CSV exercise itself. It also helps to thoroughly identify and close any gaps in the computer systems. The CSV should ensure that the organization gets the most out of it while meeting regulatory requirements [5].
Definition
- Computer System: a system with one or more PCs and related software [1].
- Computerized System: A wide range of systems including automated laboratory equipment, laboratory data management, and document management systems, but not restricted to. The computerized system comprises of the parts of hardware, software, and network, along with the regulated tasks and related paperwork [1].
- Commercial (off-the-shelf, configurable) Computerized System: Software commercially available, and whose fitness for use has been demonstrated by a broad spectrum of commercial users [1].
- In-House Developed (custom-made or bespoke) Computerized System: a system produced for a customer, specifically to order, defined set of user requirements [1].
- User Requirement Specifications (URS): portrays what the system ought to do. The client necessities contain logical, business, legitimate, administrative, safety, performance and quality parts of things the future system. The user requirements serve as the basis for the Performance Qualification (PQ) [6].
- Qualification (IQ (Installation Qualification), OQ (Operation Qualification)," and PQ (Performance Qualification): is complete and systematic testing behavior of computer system before the actual use, which directly affect the use quality of computer systems. That is, the "Qualification" is the last link of computer system quality assurance [7].
- Computerized System Validation Plan: The validation plan shall be an approved document, which describes the validation activities and responsibilities. The validation plan specifies the Computerized System subjected to validation and compiles the validation activities to be performed and the validation targets/criteria to be fulfilled. The validation plan shall be prepared and approved prior to conducting the test [8]. Black-Box Validation: Validation based on the fact that, for a given computerized system, its source code or design is unknown to the user. Validation is performed from the computerized system or computer system user’s point of view [1].
- Black-Box Test: Periodic check of a computer, computerized system or computerized system based on the black-box validation approach. Black box testing examines the functionality of a system without peering its inner structure or workings [1].
CSV Requirements
The Requirements for validation of computer systems can be found in: a. FDA 21 CFR part 820.70 b. FDA 21 CFR part 11.10 c. FDA 21 CFR part 11 d. FDA Guidance Document regarding Software Validation (also addressing process software) e. ISO 13485, clasuses 4.1.6, 7.5.2.1 and 8.2.3 f. GMP directives g. GAMP 5, e.g. regarding the "risk-based approach of testing GxP systems".
History of GAMP 5 [9]
The guidlines laid out in Good Automated Manufacturing Practices GAMP 5, for the computer qualification of automated systems including:
- Automatic computerized manufacturing equipment,
- Control systems,
- Automated laboratory systems,
- Manufacturing execution systems
- Computers running laboratory
- Database systems. The V model of GAMP 5. It is based on the standards of PQLI1, ICH Q8, ICH Q9, ICH Q10, and ASTM E2500. History of GAMP 5 explined briefly in Figure 1.
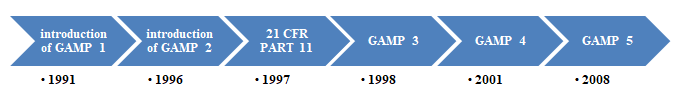
GAMP Aim
GAMP describes a set of principles and procedures that help ensure that pharmaceutical Software have required quality. Computer system validation (CSV) following GAMP guidelines require users and suppliers to work together so that responsibilities regarding the validation process are understood. A series of events has driven changes in industry standards; which will likely continue to evolve as technology advances. One of the obstacles a software development company in the Life Sciences industry routinely faces is the ability to remain current and knowledgeable on all of these developments. Being in the industry since the introduction of GAMP 1. These computerized systems generally consist of the hardware, software and netware components, together with all control functions and GAMP 5 is a useful guide in scoping your validation online activities for such systems.
GAMP from User Point of View
For users: GAMP provides a documented assurance that a system is appropriate for the intended use before it goes “live.”
GAMP from Supplier Point of View
Suppliers can use GAMP to test for avoidable defects in the supplied system to ensure quality products are produced. It must be remembered at all times that GAMP is collective ideas from the industry and does try to be all things to all people.
IT Infrastructure Control and Compliance
The GAMP® Good Practice Guide: IT Infrastructure Control and Compliance: covers a range of IT Infrastructure, from those operating globally to isolated or semi-isolated. Key aspects considered include:
- IQ, OQ of infrastructure components.
- Configuration management and change control of infrastructure components.
- Settings the infrastructure components in a highly dynamic environment.
- Management of risks to IT Infrastructure.
- Service providers fot critical infrastructure processes to be envolved.
- Security management in relation to access controls.
- Data integrity.
- Backup, restore, and disaster recovery.
- Archiving. To avoid unnecessary effort, this Guide describes a horizontal, or platform based, approach, the benefits of which include:
- Higher level of standardization throughout the entire life cycle
- Minimal overlap in documentation
- Minimal overlap in qualification This categorization covers the computerized systems, which have an impact on the Products related to patient safety, product quality, and data integrity. The categorization basically covers software and hardware Categories as explained in Tables 1 & 2. A. Software Categories
- Minimal overlap in audits, inspections, and assessments [10].
GAMP 5 Categories
| Category | Description | Validation Approach | Typical Example | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Category-1 Infrastructure Software | Layered Software Software used to manage the operating environment | Record version number, verify correct installation by following approved installation procedures. | • Operating Systems • Database Engines • Middleware • Programming Languages • Statistical Packages • Spread sheets • Network Monitoring Tools • Scheduling Tools • Version Control Tools | ||||||||
| Category-2 Firmware | No Longer Use | ||||||||||
| Category-3 Non- Configured Software | Run Time Parameters may be entered and stored, but the software cannot be configured to suit the business process | Abbreviated life cycle approach: URS, Risk- based approach to supplier assessment, Record version number, verify correct installation, Risk-based tests against requirements as directed by use. Procedures in place for maintaining compliance and fitness for intended use. | • Firmware based applications • COTS software • Laboratory Software • PLC | ||||||||
| Category-4 Configured Software | Software, often very complex, that can be configured by the user to meet the specific needs of the user’s business process. Software code is not altered | Life Cycle Approach: Risk-based approach to supplier assessment, Demonstrate supplier has adequate QMS, Some life cycle documentation retained only by supplier (e.g. Design Specification). Record Version Number Verify correct installation. Risk- based testing to demonstrate application works as designed in the test environment. Risk-based testing to demonstrate application works as designed within the business process. Procedures in place for maintaining compliance and fitness for intended use. Procedures in place for managing data. | • LIMS • Data Acquisition System • SCADA • ERP • DCS • BMS • Spreadsheets • HMI |
Table 1: Software Categories according GAMP 5. B. Hardware Categories
| Category-5 Custom software | Software Custom designed and coded to suit the business process | Same as configurable, Plus: More rigorous supplier assessment, with possible supplier audit. Full Life cycle (FS, DS, Structural Testing, etc.) Design and Source Code Review. | • Internally and Externally developed IT Applications • Internally and externally developed process control Applications. • Custom Ladder Logic. • Spreadsheets-Macro. |
|---|
Table 2: Software Categories according GAMP 5. B. Hardware Categories
| Category | Hardware type | Validation Approach | Example | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Category- 1 | Standard hardware components | Standard hardware components should be documented including make or supplier details and version number. | PLC, Controller, Scanner. | ||||||||
| Hardware details can be taken from the hardware data sheet or specification material. | |||||||||||
| Category- 2 | Custom built hardware components | Hardware should have design specification and be subjected to acceptance testing. | PCB etc. | ||||||||
| Any hardware configuration should be defined in the design documentation and verify in the IQ. |
Table 3: Hardware Categories according GAMP 5.
Importance of URS
Recent research has highlighted that in the pharmaceutical and bio-medical industry, 32% of all equipment procurement is unsatisfactory. The major problem has been identified as companies not specifying in sufficient detail and or accuracy, what their actual needs are. The lack of a fully detailed company approved User Requirements Specification (URS) , leads to many companies having to resort to otherwise un-necessary and costly retrospective actions in modifying the equipment or producing unspecified documentation or engineering drawings, post procurement. These extraneous GMP requirements often cost more than the equipment [11].
Typical Software Requirements [12]
Typical software requirements should specify the following:
- All software System inputs;
- All software System outputs;
| All functions that the software system will perform; | |
| All performance requirements that the software will | |
| meet. | |
| The definition of all external and user interfaces. | |
| How users will interact with the system; | |
| What constitutes an error and how identified errors | |
| should be handled? | |
| Required response times; | |
| The intended operating environment for the software. | |
| All ranges, limits, defaults, and specific values. | |
| All safety specifications & features. |
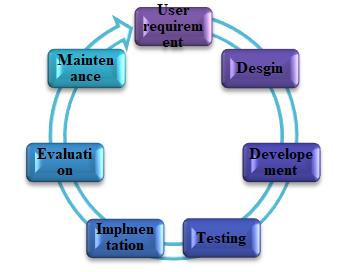
System Development Life Cycle [12]
The system development life cycle (SDLC) can be defined as, a framework for developing computer based information system. In order words, SDLC is the overall process of developing information system through a multi-step process from investigation of initial requirements through analysis, design, implementation and maintenance. These activities are carried out in different phases, which are explained in figure 2.
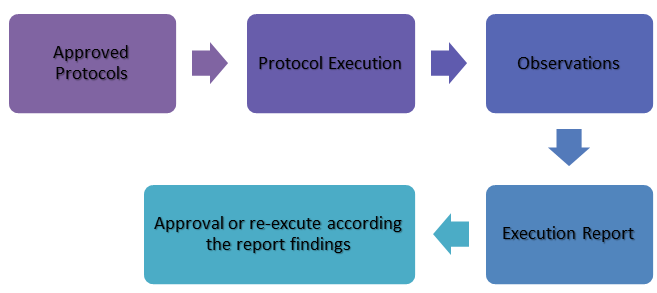
Qualification Activities [2,13]
The validation process for 21 CFR Part 11 compliance consists of these core elements:
- Comprehending the regulatory requirements.
- Ensuring compliance with CSV requirements in a cost-effective process.
- Preparing validation CSV master plan.
- Writing the CSV protocol.
- Conducting testing protocol of software and computer systems – initial and ongoing.
- Ensuring that the bare minimum documentation that FDA inspectors will ask for are available.
- Qualifying the IT systems network infrastructure and validating the network systems.
- The key activities in computer system qualification explained briefly in Figure 3.
Project Phase Deliverables
Following section provide a snapshot of project phases deliverables applicable for software validation. However, the actual deliverables to be created, reviewed and approved for a project should be identified in the project validation plan in accordance to the approach defined for the project as explained in Table 3.
| C | GAMP | 1 | GAMP | C | GAMP | C | GAMP | 5 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | .No | . | Deliverables | ||||||||||||||
| ategory- | Category-3 | ategory -4 | ategory - | ||||||||||||||
| 1. | User Requirement Specification | | | | | ||||||||||||
| 2. | Vendor Assessment | | | | | ||||||||||||
| 3. | Initial Risk Assessment | | | | | ||||||||||||
| 4. | Project Validation plan | | | | | ||||||||||||
| 5. | Functional Specification | | | | | ||||||||||||
| 6. | Functional Risk Assessment | | | | | ||||||||||||
| 7. | Configuration Specification | | | | | ||||||||||||
| 8. | Design Specification | | | | | ||||||||||||
| 9. | Setup, Configuration& Testing in validation Environment | | | | | ||||||||||||
| 10. | Installation Qualification | | | | | ||||||||||||
| 11. | Operational Qualification | | | | | ||||||||||||
| 12. | PerformanceQualification-1 | | | | | ||||||||||||
| 13. | Setup, Configuration& Testing in Production Environment | | | | | ||||||||||||
| 14. | PerformanceQualification-2 | | | | | ||||||||||||
| 15. | Traceability Matrix | | | | | ||||||||||||
| 16. | Project Validation Report | | | | |
Table 5: Project Phase Deliverables.
Compliance Strategy
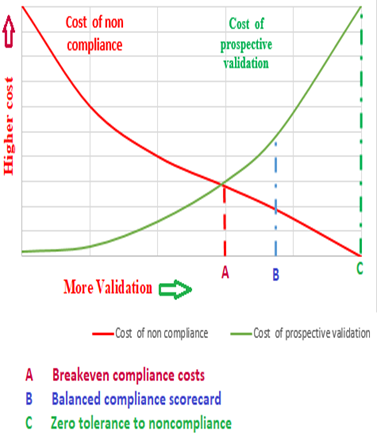
The objective must be to achieve compliance as cost- effectively as possible. Many biopharmaceutical companies have subsequently found at their expense that inefficient compliance programs are extremely costly, involving much more work than is really necessary [5].
Figure 4 illustrates three basic compliance strategies by comparing the cost associated with compliance (prospective validation) compared to the costs of non- compliance (combined effect of retrospective validation and disturbance of company).
Common Reasons for CSV Failure
Without adequate planning and preparation [5], computer system validation can encounter several problems, eventually leading to failure of the process. Problems include:
a. Inadequate documentation of plans. b. Inadequate definition of what constitutes the computer system. c. Inadequate definition of expected results. d. Inadequate specification of software. e. Software that does not meet its specifications. f. Unavailable source code for software.
Regulatory Requirements
This section identifies the Regulatory requirements, determined by following regulations [1, 14, 15]:
I. US Food & Drug Administration - Code of Federal
Regulations, Title 21, part 11: “Electronic Records; Electronic Signatures; Final Rule” II. Guide to Good Manufacturing Practice for Medicinal Products (The Rules Governing Medicinal Products in the European Community, Volume IV – Annex 11). The Regulatory Requirements are grouped according to the following Regulatory Topics [16, 17, 18, 19].
- Quality System: related to the Quality System and to the associated documentation
- Security: related to the general features of System Security and Security of Regulated Electronic Record managed by the system
- Integrity: related to the Integrity of the Regulated Electronic Record managed by the system and associated Validation documentation
- Traceability: related to the Traceability of the Regulated Electronic Record managed by the system
- Accountability: related to the Regulated Electronic Signatures managed by the system More details about the requirements in Table 4.
| 21 CFR | EU cGMP | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rule | Detailed | ||||||||||||||||
| Regulatory Topic | Part | Annex 11 | Descrption | ||||||||||||||
| Requirement | Requirement | ||||||||||||||||
| 11[14] | [1] | ||||||||||||||||
| Infrastructure Qualification | The application should be validated; IT infrastructure should be qualified. | ||||||||||||||||
| Principle | |||||||||||||||||
| Risk Management | Risk management should be applied throughout the lifecycle of the computerized system. Protocols, acceptance criteria, procedures and records based on their risk assessment. | ||||||||||||||||
| 1 | |||||||||||||||||
| Validation Standards | Standards should base on risk assessment. | ||||||||||||||||
| 4.1 | |||||||||||||||||
| System Inventory | An up to date listing of all relevant systems and their GMP functionality (inventory) should be available. | ||||||||||||||||
| 4.3 | |||||||||||||||||
| 4.5 | Supplier Qualification | The supplier should be assessed appropriately. | |||||||||||||||
| 3.2 | Quality System | ||||||||||||||||
| Quality System | 3.4 | Documentation | |||||||||||||||
| Verification | Supplier Documentation for Customized Computerized Systems | process in place should be available to ensures the formal assessment and reporting of quality and performance measures | |||||||||||||||
| 4.6 | |||||||||||||||||
| Automatic Testing Tools Adequacy | Automated testing tools and test environments should have documented assessments for their adequacy | ||||||||||||||||
| Test Environments | |||||||||||||||||
| 4.7 | |||||||||||||||||
| Incident Log | All incidents, not only system failures and data errors, should be reported and assessed. | ||||||||||||||||
| 13 | |||||||||||||||||
| Critical incident should be identified and should form the basis of CAPA. | |||||||||||||||||
| Personnel Training | All personnel should have appropriate qualifications, level of access and defined responsibilities to carry | ||||||||||||||||
| 11.10 | |||||||||||||||||
| 2 | |||||||||||||||||
| (i) | |||||||||||||||||
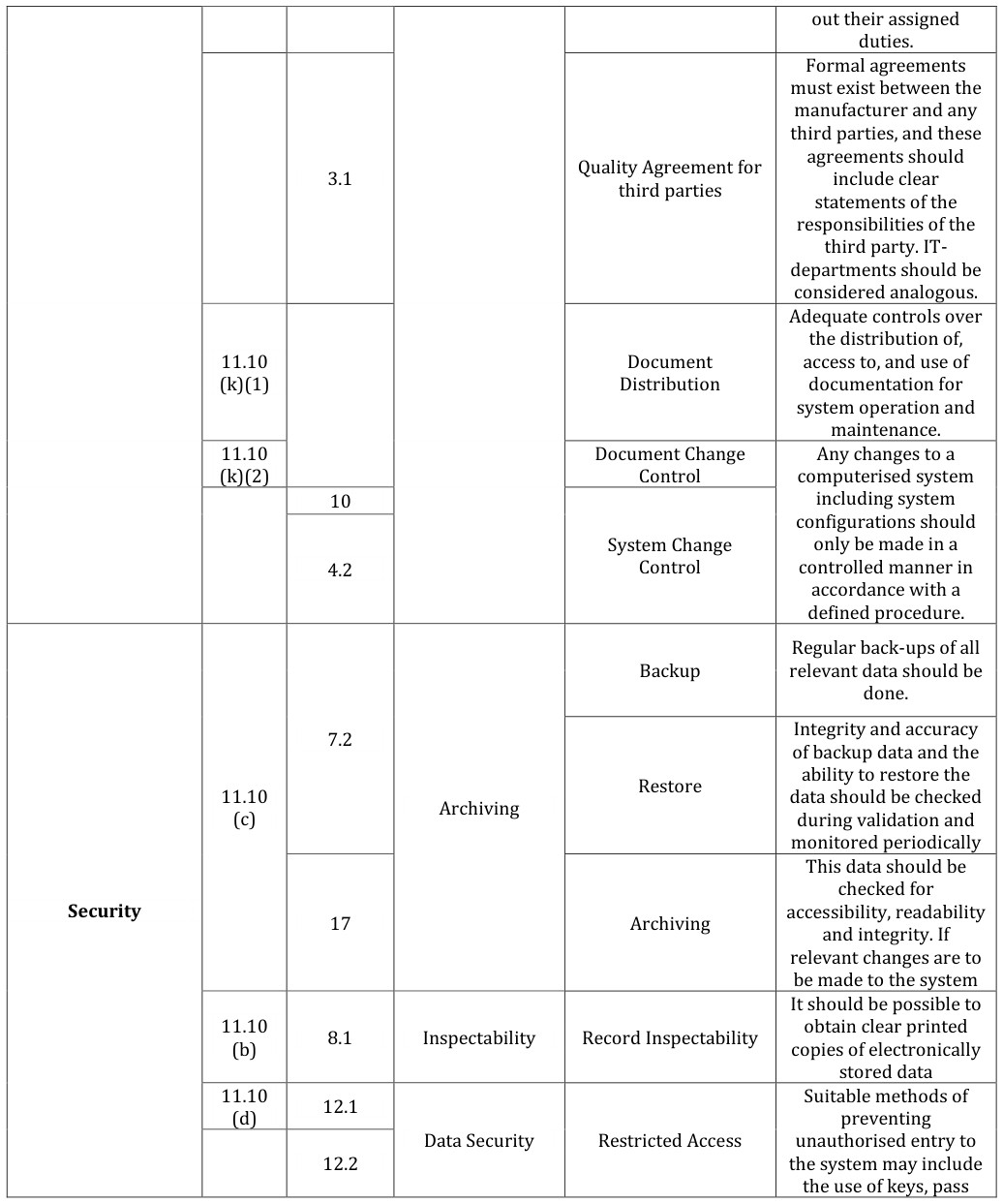
11.300 (a) 12.1 Uniqueness of Codes
11.10 (g) 12.1
Integrity 11.10 (a) cards, personal codes with passwords, biometrics, restricted access to computer equipment and data storage areas safeguards in place to prevent unauthorized use of passwords and/or identification codes, The system can detecr and report in an immediate and urgent manner any attempts at Authority Check their unauthorized use to the system security unit Automatically log out Automatic Log Off users after a defined period of inactivity Users should work only under their own user profiles encompassing unique user IDs and individual passwords or User Profiles Security other access keys and not share these with othersCreation, change, and cancellation of access authorizations should be recorded.
Data should be secured
by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period.
Decisions on the extent
of validation and data integrity controls should be based on a justified and documented risk assessment of the computerized system. 11 Periodic Review Computerized systems
4.7 Validation Testing
4.8 Data Migration
6 Invalid Records Invalid Records
Altered Record Altered Record
16 Business Continuity Business Continuity
should be periodically evaluated to confirm that they remain valid.
URS should describe the required functions of the computerized system and be based on
4.4 User Requirements
documented risk assessment and GMP
impact.
An up to date listing of all relevant systems and
4.3 System Specifications
their GMP functionality
(inventory) should be available Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered.
If data are transferred to another data format or system, validation should include checks Verification that data are not altered in value and/or meaning For critical data entered manually, there should be an additional check Detection on the accuracy of the data.
Validation of systems to
ensure accuracy, reliability, consistent intended performance, Detection
and the ability to discern invalid or altered records availability of computerized systems supporting critical processes, provisions should be made to ensure continuity of support for those processes in the event of a system breakdown
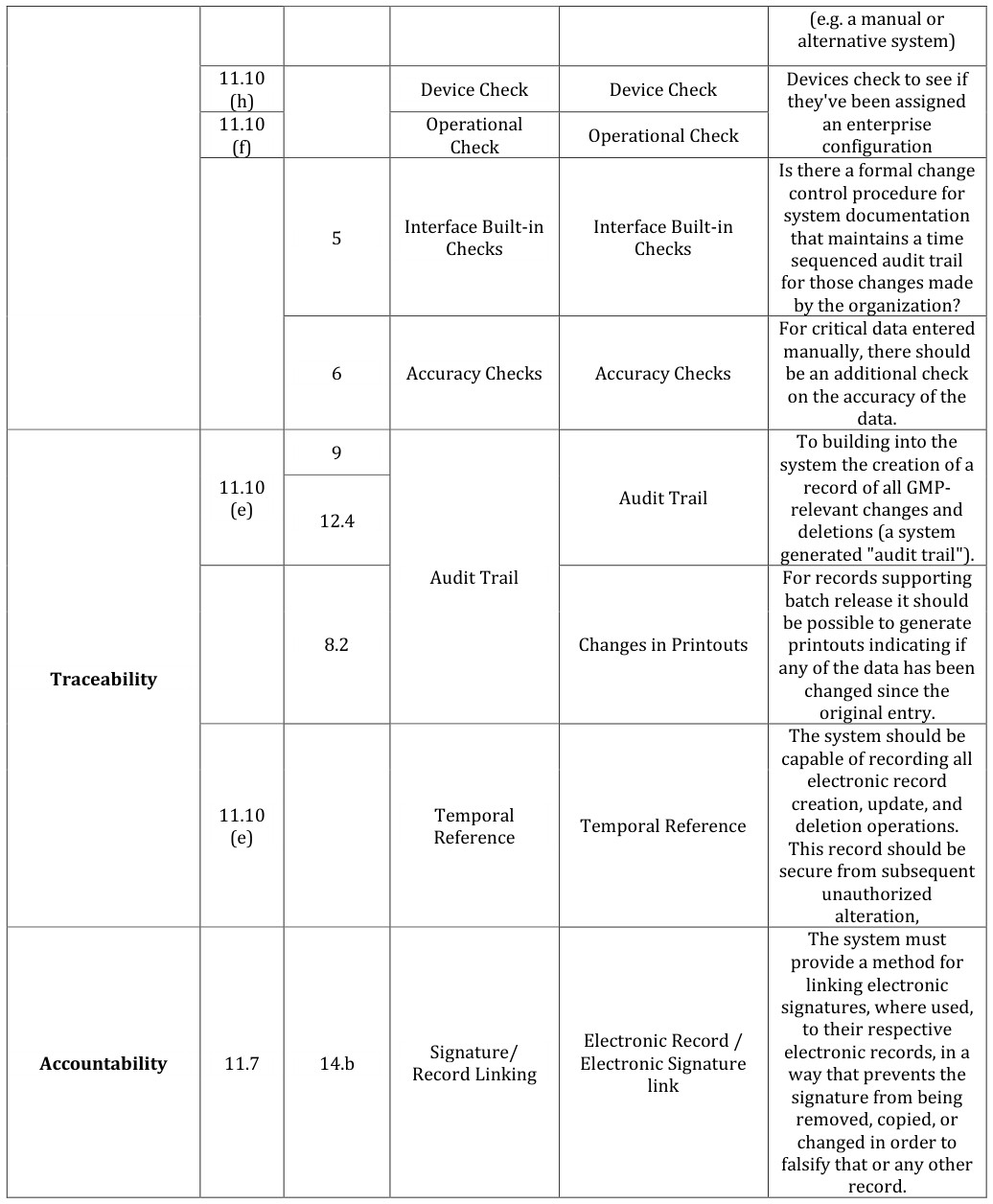
Device Check Device Check Devices check to see if that maintains a time sequenced audit trail for those changes made To building into the system the creation of a batch release it should be possible to generate printouts indicating if any of the data has been The system should be capable of recording all deletion operations. This record should be secure from subsequent changed in order to falsify that or any other
11.3 (a,b,d) Physical and/or logical
controls should be in place to restrict access to computerized system Uniqueness of identification Components (i.e. code to authorized persons.
The extent of security controls depends on the and password) criticality of the computerized system.
Initial and periodic testing of devices, such as tokens or cards, that
- 12
- Electronic
- Signature
- Management
- 14.a
- Electronic Signature
- 4.1
- Hybrid Management
- Electronic
- 11.50 (a)
- 14.c
- Signature
- Manifestation
- 15
- Batch Release
- Batch Release and QP
Table 7: 21 CFR Part 11, EU cGMP Annex 11 requirements for CSV.
system requires validation to be comply GMP requirements, Examples for these systems mentioned in table 5.
Examples from Computerized System Required CSV
In biopharmaceutical industries many computerized
| S. No. | Examples for computerized system required CSV | Risk on GMP or Business | ||||||
|---|---|---|---|---|---|---|---|---|
| 1.1 | Operation system software | Business | ||||||
| 1.2 | Servers and backup solution software | Business & GMP | ||||||
| 1.3 | Documents related software | Business & GMP | ||||||
| 1.4 | Software for materials stock control | Business & GMP | ||||||
| 1.5 | Software or excel sheets related to materials / batches release | Business & GMP | ||||||
| 1.6 | Software for autoclave operation | Business & GMP | ||||||
| 1.7 | Identification IDs Printer software | GMP | ||||||
| 1.8 | Fermentations operation software | Business & GMP | ||||||
| 1.9 | Filter integrity tester software | GMP | ||||||
| 1.10 | Inoculator / Harvester Operation Software | GMP | ||||||
| 1.11 | Washing / Sterilization Tunnel operation software | GMP | ||||||
| 1.12 | Blending system operation software | GMP | ||||||
| 1.13 | Filling line operation software | GMP | ||||||
| 1.14 | Labeling / Batch details printing software | Business & GMP | ||||||
| 1.15 | Thermal mapping software | GMP | ||||||
| 1.16 | BMS software | Business & GMP |
Table 8: Examples for computerized systems requires CSV.
Conclusion
Without adequate planning and preparation, computer system validation can encounter several problems, eventually leading to failure of the process so the successful computer system validation (CSV) is highly dependent upon the quality assurance system, a formal System Development Life Cycle, and the qualification tasks performed throughout the this cycle. CSV must establish a level of confidence‖ that the system consistently meets the requirements and user requirements. As most methodologies require that specifications and test protocols are written, approved by qualified staff, and acted upon, it is possible to adapt the validation methodology to most situations, provided that the system requirements and functionality can be shown to be tested and proven, and that the system development, implementation, and operation is under control. Above all the system must be shown to operate correctly. Above all, the device must be shown to function properly, reliably and in compliance with its requirements. The system must be validated according the quality system and approved protocols to provide the user by data integrity, security, traceability and accountability.
Acknowledgment
Many thanks for Dr. Momtaz Wasfy for his revision and his generously contributed to the work presented in this review. In particular, Eng. Amr Mostafa for their tremendous support, but also for giving me many wonderful opportunities to learn many amazing things from them.
References
-
Union E (2011) EudraLex The Rules Governing Medicinal Products in the European Union - Volume 4 Good Manufacturing Practice. Medicinal Products for Human and Veterinary Use - Annex 11: Computerised Systems, pp: 1-90.
-
Ostrove SA (2016) How to Validate a Pharmaceutical Process How to Validate a Pharmaceutical Process.
-
(2019) Computer System Validation in the Pharma Laboratory - 10 years of GAMP 5, Pitfalls and Best Practices.
-
Rodríguez-Pérez J (2014) The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished Pharmaceuticals.
-
Wingate G (2010) Pharmaceutical Computer Systems Validation Quality Assurance, Risk Management and Regulatory Compliance 2nd (Edn.).
-
Zwetkow M, Tanguay S (2013) Qualification Guideline for Microsoft Office 365. Heal Life Sci Ind Unit Microsoft.
-
(2017) Validation and Automated Validation. Tracelink.
-
Signature E (2017) Current Practices of system validation.
-
Shields S (2013) GAMP 5 A Risk-Based Risk Based Approach to Compliant GxP Computerized Systems. Allergan, pp: 1-29.
-
(2005) GAMP Good Practice Guide: Testing of GxP Systems. ISPE, pp: 1-166.
-
Lead G (2018) Guidelines on Validation - Appendix 5 Validation of Computerized Systems. World Health Organization, pp: 1-29.
-
(2002) U.S. Department Of Health and Human Services Food and Drug Administration and C. for D. and R. H. C. for B. E. and Research, General Principles of Software Validation ; Final Guidance for Industry and FDA Staff.
-
Elser C, Richmond FJ (2018) Validation Master Plans : Progress of Implementation in the Pharmaceutical Industry. Therapeutic Innovation & Regulatory Science 53(3): 354-363.
-
Orlando López (2005) 21 CFR Part 11: Complete Guide to International Computer Validation Compliance for the Pharmaceutical Industry. Boca Raton London New York Washington, DC, pp: 1-286.
-
Global compliance panel training. Your Gateway to Regulatory Compliance.
-
http://globalcompliancepanel.viewpage.co/Validatio n-and-21-CFR-11-Compliance
-
Phan TT (2003) Technical considerations for the validation of electronic spreadsheets for complying with 21 CFR Part 11. Pharmaceutical Technology North America 27(1): 50-56.
-
McDowall RD (2019) Data Integrity Focus, Part 1: Understanding the Scope of Data Integrity. LCGC North America 37(1): 44-51.
-
McDowall RD (2019) Data Integrity Focus, Part III: What Is the Problem with Hybrid Systems . LCGC North America 37(3): 180-184.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State