Current Trends in Planning, Preparation and Delivering Regulatory Submissions in India & USA
The drug markets on the planet have various prerequisites for the enrolment of a drug item. To fit the prerequisites according to the administrative organizations, an idea of basic specialized report was actualized by ICH. Getting ready administrative entries in the drug business can be an extremely upsetting encounter because of the enormous measure of work and the need to explore and comprehend administrative accommodation necessities. This paper means to help administrative experts by giving data and exhortation to arranging and setting up an administrative accommodation. The paper centres around the conveyance of accommodation information bundles, since these are commonly the duty of administrative experts. The paper will cover the Regulatory necessities for entries, Components of accommodation information bundles, arranging an accommodation and best practices for getting ready accommodation information bundles.
Introduction
This paper gives data to administrative experts to assist them with bettering arrangement and get ready entries in INDIA and USA. The paper will initially give an elevated level outline of Regulatory specialists of INDIA and USA Name Regulatory Authority Name Logo INDIA Central Drug Standard Control Organization (CDSCO) United States of America (USA) United States Food and Drug Administration (USFDA) administrative prerequisites, and parts of accommodation information bundles. The paper will at that point centre around methodologies and best practices for arranging and planning entries [1].
INDIA (CDSCO)
The Drug and Cosmetic Act 1940 and Rules 1945 were passed by the India’s parliament to direct the import, production, dispersion and offer of medications and beautifiers. The Central Drugs Standard Control Organization (CDSCO), and the workplace of its chief the Drugs Controller General (India) [DCGI] was set up. In 1988, the Indian government added Schedule Y to the Drug and Cosmetics Rules 1945. Schedule Y gives the rules and prerequisites to clinical preliminaries, which was additionally re-examined in 2005 to bring it at standard with globally acknowledged method. The progression incorporates, building up definitions for Phase I-IV preliminaries and clear duties regarding examiners and supports. Showing of wellbeing and viability of the medication item for use in people is basic before the medication item can be endorsed for import or assembling and promoting in the nation. The Rules 122A, 122B and 122D, 122 DA, 122DAA, 122E of Drugs and Cosmetics Rules and Appendix I, IA and VI of Schedule Y, depict the data/information needed for endorsement of clinical preliminary and additionally to import or produce of new medication for promoting in the nation. In any case, the prerequisites for endorsement of clinical preliminaries and new medications may fluctuate contingent upon nature of new medications.
This direction reports has been set up to indicate the overall necessities for endorsement of clinical preliminary and various classifications of New Drugs viz. Investigational New Drugs, New medications substances, extra quality, extra sign, altered delivery structure and so on This direction will assist the business with presenting the necessary records in a more practical way, which thusly will likewise help analyst of CDSCO to survey such application in orderly way. It is evident that this organized application with far reaching and sane substance will assist the CDSCO with exploring and take essential activities in a superior manner and would likewise facilitate the arrangement of electronic entries, which may occur sooner rather than later at CENTRAL DRUGS STANDARD CONTROL ORGANIZATION (CDSCO) [2].
USA (USFDA)
The U S. Food and Drug Administration is a logical, administrative, and general wellbeing office that supervises things representing 20 pennies of each dollar spent by buyers. Its locale includes most food items (other than meat and poultry), human and creature drugs, helpful specialists of organic root, clinical gadgets, radiation-transmitting items for customer, clinical, and word related use, beautifying agents, and creature feed. Organization researchers assess applications for new human medications and biologics, complex clinical gadgets, food and shading added substances, baby equations, and creature drugs. Additionally, the FDA screens the assembling, import, transport, stockpiling, and offer of about $1 trillion worth of items yearly at an expense to citizens of about $3 per individual. Agents and assessors visit in excess of 16,000 offices every year, and mastermind with state governments to help increment the quantity of offices checked [3].
The Food and Drug Administration (FDA) is an organization inside the U.S. Branch of Health and Human Services. It comprises of the Office of the Commissioner and four directorates supervising the centre elements of the office: Medical Products and Tobacco, Foods and Veterinary Medicine, Global Regulatory Operations and Policy, and Operations. FDA is liable for:
- Protecting the general wellbeing by guaranteeing that nourishments are sheltered, healthy, clean and appropriately marked; human and veterinary medications, and antibodies and other natural items and clinical gadgets planned for human use are protected and viable.
- Protecting general society from electronic item radiation.
- Assuring beautifying agents and dietary enhancements are sheltered and appropriately marked.
- Regulating tobacco items.
- Advancing the general wellbeing by assisting with speeding item advancements.
- Helping general society get the exact science-based data they have to utilize prescriptions, gadgets, and nourishments to improve their wellbeing [4]. FDA’s obligations stretch out to the 50 United States, the District of Columbia, Puerto Rico, Guam, the Virgin Islands, American Samoa, and different U.S. domains and assets.
Administrative Requirements for Submissions
One of the most significant administrative prerequisites is consistence with the Common Technical Document (CTD) standard. The CTD is a lot of determinations for entries to administrative organizations. The CTD standard was made by the International Conference on Harmonization (ICH), an association dispatched in 1990 with the objective of fitting drug administrative necessities across various nations. The CTD standard determines:
- What archives ought to be remembered for administrative filings (for example Conventions, Clinical Study Reports, Risk Management Plans, and so on)
- Specifications on the substance and configuration of numerous archives (from DIA introduction)
- Associated report type-definitions (DTDs) and templates.
- The envelope structure for putting away records and the naming shows for organizers and documents.
• Technical determinations for a XML spine record giving metadata about substance documents. The accompanying outlines give a graphical perspective on a portion of the prerequisites for the association of records and archives inside the Common Technical Document [5].
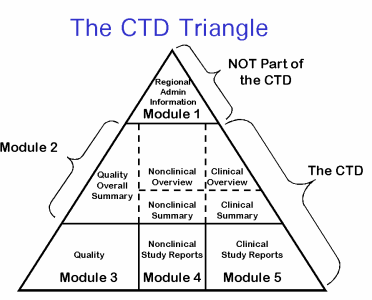
The Food and Drug Administration (FDA), European Medicines Agency (EMA), and Central Drug Standard Control Organization (CDSCO) expect consistence to CTD norms for entries, and other administrative offices are in different conditions of availability for tolerating CTD- consistent entries. Notwithstanding, organizations may have increments to the ICH’s CTD details. In this manner, it is imperative to talk with individual administrative offices to comprehend the particular CTD necessities of every office.
CTD is basic specialized archives that might be a significant venture of the ICH to maintain a strategic distance from the duplication and interpretation into provincial language work of one application. Through this, a candidate can record one single application to more than each nation in turn for the enlistment of their medication item.
Outline on CTD
There were bunches of issues happen for the interpretation as per the territorial language of an equivalent application and are commonly very tedious cycle. To conquer such issues, an idea of ICH .was pronounced in 1990. This was proclaimed by the collaboration of the three areas US, EU and Japan; which are known as three sided districts of the ICH. ICH was sorted out with a significant target to draft, affirm and execution of the rules which are acknowledged all through the three sided locales. Such rules are made covering all the perspectives and boundaries identified with the medication items for example virtue, quality, wellbeing, and adequacy perspective [6].
Methodology
Medication Approval Process In India
The Drug and Cosmetic Act 1940 and Rules 1945 were announced by the India’s parliament to manage the import, production, conveyance and offer of medications and beautifiers. The Central Drugs Standard Control Organization (CDSCO) and the workplace of its chief, the Drugs Controller General (DCGI) was set up. In 1988, the Indian government added Schedule Y to the Drug and Cosmetics Rules 1945. Timetable Y gives the rules and prerequisites to clinical preliminaries, which was additionally re-examined in 2005 to bring it at standard with globally acknowledged system. At the point when an organization in India needs to fabricate/ import another medication it needs to apply to look for authorization from the permitting authority (DCGI) by recording in Form 44 additionally presenting the information as given in Schedule Y of Drugs and Cosmetics Act 1940 and Rules 1945. So as to demonstrate its viability and wellbeing in Indian populace it needs to direct clinical preliminaries as per the rules indicated in Schedule Y and present the report of such clinical preliminaries in determined arrangement [7].
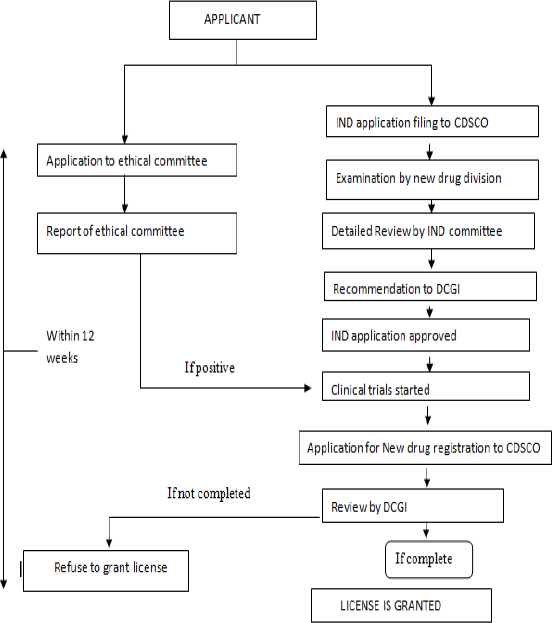
Medication Approval in United States
The United States has maybe the world’s most rigid principles for endorsing new medications. Medication endorsement guidelines in the United States are considered by numerous individuals to be the most requesting on the planet. The essential guideline can be perceived.
Sorts of Applications Investigational New Drug (IND): It’s an application documented to the FDA so as to begin clinical preliminaries in people if the medication was discovered to be sheltered from the reports of Preclinical preliminaries. A firm or foundation, called a Sponsor, is liable for presenting the IND application [4]. A pre-IND meeting can be masterminded with the FDA to examine various issues [8]:
- The plan of creature research, which is needed to loan backing to the clinical examinations
- The planned convention for leading the clinical Trial [8].
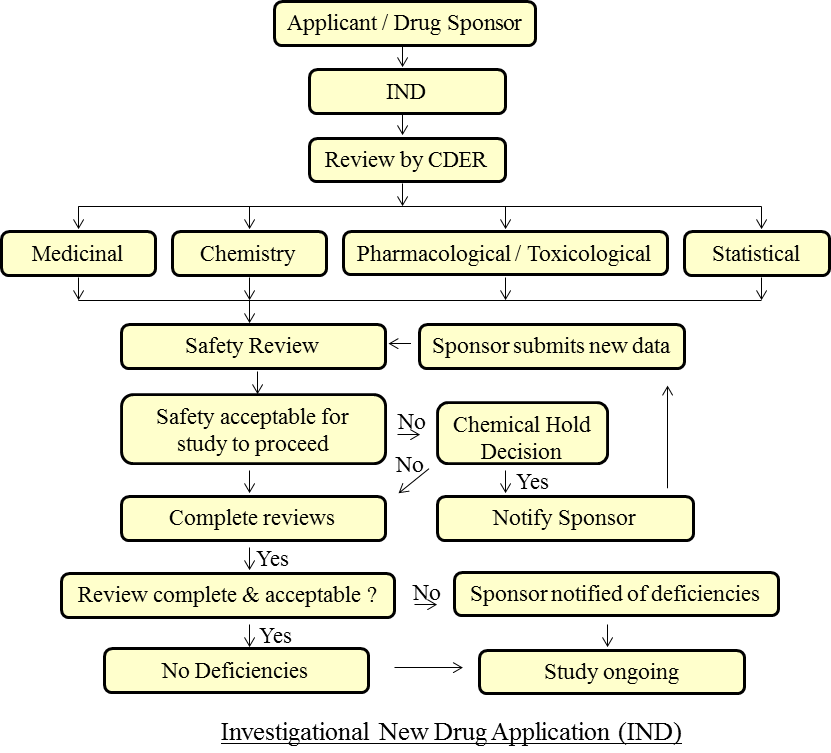
There are three IND types:
- An Investigator IND is presented by a doctor who the two starts and leads an examination, and under whose quick heading the investigational drug is controlled or apportioned. A doctor may present an exploration IND to propose contemplating an unapproved drug, or an endorsed item for another sign or in another patient populace.
- Emergency Use IND permits the FDA to approve utilization of an exploratory medication in a crisis circumstance that doesn’t permit time for accommodation of an IND as per 21CFR.
- Treatment IND is submitted for trial drugs indicating guarantee in clinical testing for genuine or quickly hazardous conditions while the last clinical work is led and the FDA audit happens [9].
New Drug Application (NDA): At the point when the backer of another medication accepts that enough proof on the medication’s security and adequacy has been gotten to meet FDA’s prerequisites for advertising endorsement, the support submits to FDA another medication application (NDA). The application must contain information from explicit specialized perspectives for audit, including science, pharmacology, clinical, bio pharmaceutics, and insights. On the off chance that the NDA is affirmed, the item might be advertised in the United States [10].
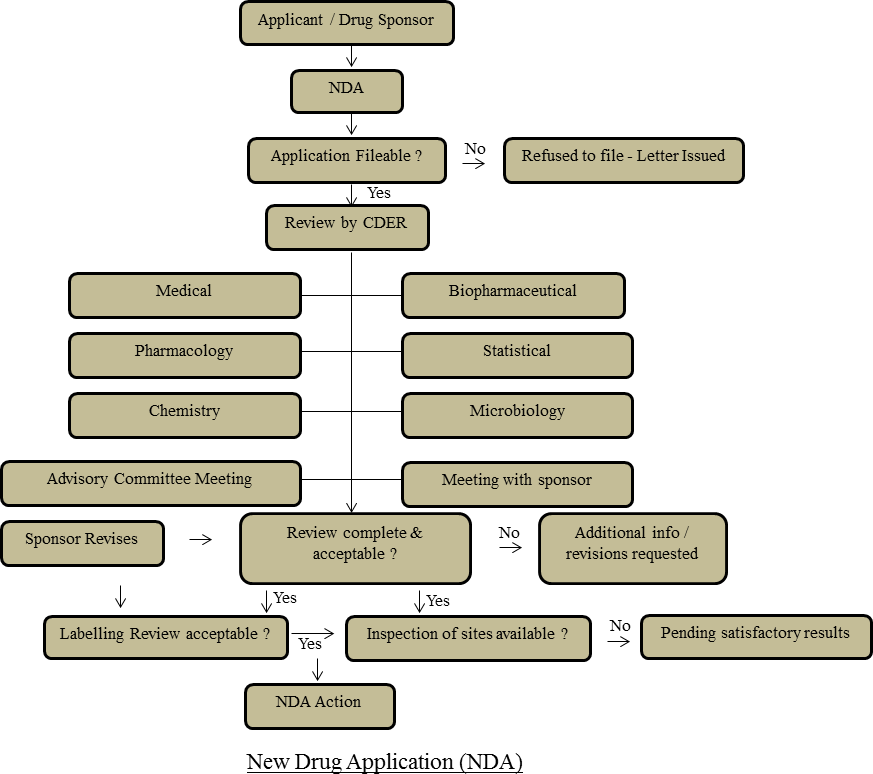
Laws, Regulations, Policies and Procedures: The mission of FDA is to authorize laws ordered by the U.S. Congress and guidelines set up by the Agency to ensure the purchaser’s wellbeing, security, and wallet. The Federal Food, Drug, and Cosmetic Act is the essential food and medication law of the U.S. The law is expected to guarantee shoppers that nourishments are unadulterated and healthy, safe to eat, and delivered under clean conditions; that medications and gadgets are sheltered and successful for their planned uses; that beautifiers are protected and produced using fitting fixings; and that all naming and bundling is honest, instructive, and not beguiling [11].
Code of Federal Regulations (CFR): The last guidelines distributed in the Federal Register (day by day distributed record of proposed rules, last principles, meeting sees, and so on) are gathered in the Code Of Federal Regulations (CFR). The CFR is isolated into 50 titles that speak to wide territories subject to Federal guidelines. The FDA’s bit of the CFR deciphers The Federal Food, Drug, and Cosmetic Act and related rules. Area 21 of the CFR contains most guidelines relating to food and medications. The guidelines archive all activities of all medication supports that are needed under Federal law [12].
Results and Discussion
| Requirements | INDIA | USA |
|---|---|---|
| changes | Changes: • Major • Moderate | changes in the approved drug: • Minor • Moderate • Major |
| Application | MAA | ANDA/NDA |
| Debarment classification | Not required | Required |
| Number of copies | 1 | 3 |
| Approval timeline | 2 - 18 months | 18 months |
| Presentation | Paper | eCTD and Paper |
| Number of batches | 1 | 1 |
| Packaging | Not addressed | A minimum of 1,00,0000 |
| Process validation | Required | Not required at the time of submission |
| Batch size | Pilot scale batch | 1 pilot scale or minimum of 1 lakh units whichever is higher. |
Table 1: Comparison of Regulatory requirements between INDIA and USA.
Summary and Conclusion
Planning Regulatory accommodation information bundles is difficult work. Entries require numerous examinations, datasets, projects, and records. A conscious and proactive methodology is vital to staying away from a minute ago scrambles to comply with time constraints or to make changes to meet beforehand obscure prerequisites. Best practices for a smooth administrative accommodation include:
- Start arranging early
- Educate yourself on guidelines and parts of entries
- Work intimately with all administrative partners to proactively look for early concurrence with administrative organizations
- Create a hearty venture the executive’s plan that incorporates all segments
- Build in quality from the earliest starting point
- Regulatory entries are a territory where an early venture of time, training, and arranging will pay off with time and asset investment funds over the long haul.
CTD is the basic organization for documenting dossiers. To set up a cycle by which eCTD accommodation can be considered on a normal premise in future. There should be a framework to create xml spine. Source records and information should be submitted ought to be prepared and consistent with necessities and certain focuses that may determine to be in consistence ought to be viewed as like setting guidelines before all else, picking important document names, keeping comparable examinations together, clear correspondence with analysts, picking a fitting degree of granularity, applying supportive hyperlinks, considering future life cycle the board and so on Supporters ought to comprehend, know and solid and steady on how organizations utilize the segments of an CTD for survey.
Acknowledgment
We express our deepest gratitude to the Management of Vision College of Pharmaceutical Sciences and Research for timely help and co-operation to the pillars of the research work.
Conflict of Interest
The authors declare no conflicts of interest relevant to this article.
References
-
Dureja H, New medication endorsement measure in India.
-
Horner A (2005) Comparison of a worldwide accommodation of new organic substance and another compound element - key choices and models for usage.
-
Kurian G (1998) A Historical Guide to the U.S. Government. Oxford university press, UK, pp: 768.
-
Parker R (2005) FDA Administrative Enforcement Manual. 1st (Edn.), CRC press, pp: 220-221.
-
Nordfjeld K, Strasberger V (2006) Creating eCTD applications. Journal of Generic Medicines 3(2): 140-146.
-
Powell S (2007) A Successful Transition to CTD and eCTD. Regulatory Affairs Journal Pharma.
-
Berry IR, Martin RP (2008) The Pharmaceutical Regulatory Process. 2nd (Edn.), CRC press, pp: 48.
-
U.S. Food and medication organization, Investigation of new medication.
-
Morris D (2008) LLP: OIG Reports on FDA Generic Drug Review Process.
-
Guarino RA New medication endorsement measure. 3rd (Edn.), Marcel Dekker distribution, New York, pp: 69-70.
-
Monahan C, Babiarz JC (2008) The new drug application. In: Pisano DJ, et al. (Eds.), FDA Regulatory issues. A guide for physician recommended drugs, clinical gadgets, and biologics. 2nd (Edn.), CRC press, New York.
-
Silverstein B (2011) Reveal to me a story: Writing a Persuasive Regulatory Document. Administrative Focus, pp: 35-37.
- Hydrogen Peroxide Scavenging by Methanolic Extracts of Coriander: An In Vitro Antioxidant Study
- Aromatherapy in Palliative Care: A Fragrant Quest for Relief
- Empowering Women, Securing Futures: Contraception’s Role in Socioeconomic Progress in India
- Effect of Crospovidone, Croscaramellose Sodium in Combination on the Drug Release of Anti diabetic Medication in Tablet Form
- Knowledge, Attitudes, Anxiety, and Preventive Behaviors Regarding Covid-19 Affliction among Healthcare Workers in Pakistan
- “Competitive Landscape and Brand Equivalents: Implications for ANDA (Abbreviated New Drug Application) Approval”