Could UPR Manipulation Help to Tune the Inflammatory Response in the Course of COVID-19?
Highly pathogenic coronavirus SARS-CoV2, belonging to coronaviridae family, preferentially infects alveolar epithelial cells and immune cells resident or recruited in the lung, causing the disease known as COVID-19. As for other viruses, SARS-CoV2is sensed by several PRRs, particularly TLR3 that triggers an intracellular signaling culminating in activation of transcription factors that promote the release of inflammatory and anti-viral cytokines, deeply shaping immune response. In a subgroup of patents, a massive release of inflammatory cytokines may also occur, strongly contributing to destroy alveolar cells, fibrosis and endothelial injury, thus favoring the activation of coagulation cascade. Viral infection also triggers UPR, an integrated response to stress, by activating the antiviral kinase PKR and by perturbing ER homeostasis. ER stress/UPR strongly contributes to the regulation of cytokine release also because its signaling intersects with PRR signaling at multiple levels. In this perspective we will discuss the possibility to tune the inflammatory/immune response to SARS-CoV2 infection by reducing ER stress, manipulating the different arms of UPR or inducing autophagy.
Perspective
Viruses infecting target cells via specific receptor/co- receptor molecules are sensed by PRRs including TLRs, NODs, C-type lectin receptors and RIGs, located in different cellular compartments. PRRs bind pathogen components (PAMPs) with a partial specificity, recruit adaptive molecules such as Myd88 and TRIF and trigger an intracellular signaling leading to the phosphorylation of transcription factors such as NFkB, MAPKs, AP-1 and IRFs [1]. Phosphorylation results in their activation and translocation into the nucleus where they promote the transcription of anti-viral, pro-inflammatory and anti-inflammatory cytokines, deeply shaping immune response [2]. Viral infection may induce tissue damage exposing/releasing DAMPs such as HMGB1 or HSPs that may be also sensed by PRRs, amplifying inflammatory response.
Coronaviridae family encompasses a group of positive- Perspective
sense single-strand RNA viruses that include the high pathogenic respiratory viruses SARS-CoV and SARS-CoV-2 that causes Coronavirus disease 2019 (COVID-19). These viruses mainly infect airway epithelial cells and immune cells such as alveolar macrophages located at the interface between the environment and host, using as main receptor angiotensin converting enzyme (ACE) 2 [3]. TLR3, expressed by alveolar and bronchial epithelial cells as well as by immune cells resident or recruited in the lung, is mainly involved in the activation of in response to coronavirus infection. TLR signaling activates NFkB and IRFs lading to the production of pro-inflammatory and anti-viral cytokines even if the highly pathogenic coronaviruses are poor type I IFN inducers [4, 5]. Therefore the use of TLR3 agonists such as PolyIC could be helpful to potentiate the anti-viral response. Moreover, TLR4 is up-regulated in alveolar epithelial cells following coronavirus infection and its binding to HMGB1 amplifies inflammation that could be controlled by TLR4 antagonists such as Eritoran. Besides PRRs, viral infection triggers UPR, an integrated stress response initiated by the activation of the antiviral kinase PKR and/or by the accumulation into the ER of unfolded/misfolded viral proteins that perturb ER homeostasis [6]. UPR is orchestrated by three main sensors namely IRE1α PERK and ATF6 that, in not stressful conditions, are inactive due to their binding to GRP78/BIP. Unfolded/misfolded proteins in the ER attract BIP, detaching it from UPR sensors resulting in their activation. Unless is too strong or too long, UPR helps cells to adapt to stress, i.e. PERK-eIF2α axis reduces protein translation, IRE1α increases ER chaperone transcription, mRNA degradation via the IRE1α-RIDD axis and protein catabolism via ERAD or via macro autophagy, although the activation of the latter process contribute the other two branches of UPR [7].
ER stress may be also induced because viruses interfere with the functions of the ER as part of their infectious life cycle, as it has been reported to occur in the course of SARS-CoV that induces ER reorganization, leading to the formation double membrane vesicles (DMVs) that are closely associated to the viral replication/transcription complexes (RTCs) [8]. Last but not least, viruses may induce ER stress by interfering with macro autophagy, more often at the final steps of the process, to avoid their elimination [9]. This strategy has been reported to be utilized by MERS- CoV that, by increasing SKP2 phosphorylation, reduced autophagosome/lysosome fusion [10]. As for autophagy, viruses may also manipulate UPR to their own purpose, i.e. HSV1 may restore protein translation by dephosphorylating eIF2alpha or Japanese encephalitis virus (JEV) exclude from IRE1a-RIDD- mediated RNA degradation its own RNA [11]. As viruses often infect cells of the immune system, the dysregulation of UPR may lead to immune dysfunction, being UPR involved in the differentiation/homeostasis of immune cells [12, 13]. UPR plays a pivotal role in the control ROS level, indeed protein accumulation into the ER and the formation of intermolecular and intramolecular disulfide bonds during their folding generates ROS, partially counteracted by the activation the PERK-NRF2 axis [14]. More importantly, UPR activates the most important transcription factors that regulate cytokine production, i.e. activated IRE1α binds TNF receptor- associated factor 2 (TRAF2) to phosphorylate IkB and activate NFkB, ATF6 activates NFkB through the phosphorylation of AKT and PERK-eIF2α axis, by inhibiting protein translation, reduces the expression level of IkB NFkB inhibitor that requires continuous synthesis to maintain the steady state and restrain NFkB activation [15]. Interestingly, UPR intersects with PRR signaling at multiple levels to regulate the production of cytokines in turn to re-activate UPR (Figure 1), in a positive feed-back loop, amplifying inflammation [16].
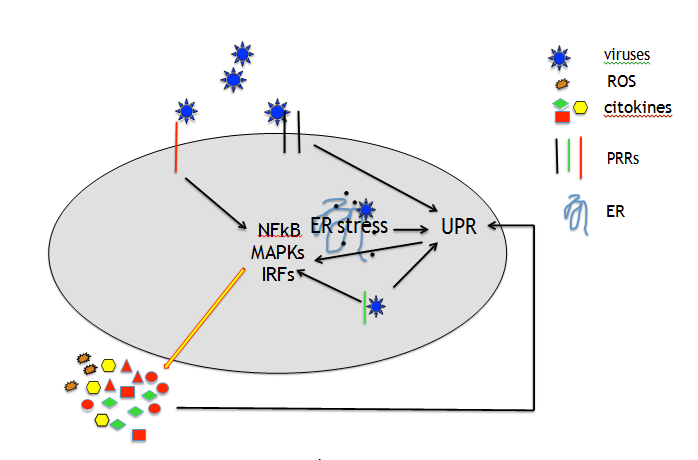
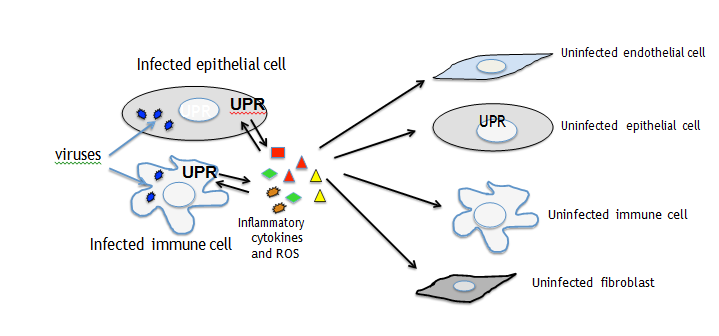
UPR may be triggered by viruses through the TLR, NOD or RIG signaling. For example the engagement of TLR2 and TLR4 may activate IRE1a through TRAF6 and NADPH oxidase 2 (NOX2) and GADD34, that de-phosphorylates eIF2α to restore protein translation, besides by PERK-eIF2α−ATF4 axis may be up-regulated by TLR and RLR signaling, through IRF3 and IRF7. Of note also MAPKs including JNK, P38 and ERK1/2, strongly involved in inflammatory response, can be activated by both PRR and UPR signaling [17]. Last but not least, UPR contributes to inflammation, by triggering NLRP3 inflammosome assembly, promoting IL-1beta and IL-18 release, in correlation with ROS production by mitochondria stressed by IRE1α activation. Although pro-inflammatory cytokines are produced as a defense mechanism against invading viruses, their massive release strongly contributes to tissue damage. Particularly, following acute infection by highly pathogenic coronaviruses such as the new emerging SARs-CoV2, in a subgroup of patients the “cytokine storm syndrome” occurs, characterized by a huge amount of pro-inflammatory cytokines such as IL-6, TNF alpha, IL-8 and CCL-2 that destroy alveolar cells, promoting the acute respiratory distress syndrome (ARDS). These cytokines, released by infected cells may mediate a cross-talk between them and bystander uninfected cells and transfer the stress and activate UPR in macrophages or dendritic cells (DCs), impairing their function. Previous studies have indeed demonstrated that UPR is activated in M2 polarized macrophages or in dysfunctional DCs exposed to the tumor microenvironment [18, 19]. Inflammatory cytokines may also damage endothelial cells inducing the release of soluble tissue factor activating the coagulation cascade or trigger UPR in fibroblasts, stimulating their trans-differentiation into my fibroblasts that promote fibrosis (Figure 2) [20, 21].
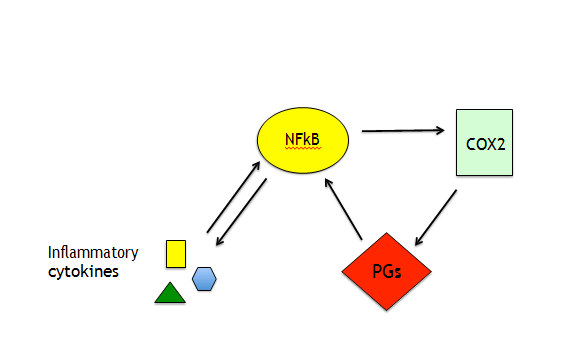
As pro-inflammatory cytokines present in the BAL are also detectable in the plasma of ARDS-affected patients they may damage other organs and also trigger the severe disseminated intravascular coagulation (DIC) to which, besides the exposure/release of tissue factor, contributes the impairment of fibrin degradation [22]. Of note, PERK-ATF4- CHOP axis of UPR promotes the release of prostaglandins (PGs) generated by cyclooxygenases (COXs). They contribute to cytokine transcription by activating NFkB that in turn activates COX2 to further produce PGs (Figure 3). These molecules, particularly PGE2, besides inflammation, reduce anti-viral immune response by inhibiting IFN and nitrogen oxide (NO) production and may also favor viral replication [23]. This suggests that the use of COX2 inhibitors could also be promising drugs in the treatments of SARS- CoV-2-infected patients. One of the pathways involved in immune dysfunction activated by cytokines through Janus kinases (JAKs) is STAT3 that is also phosphorylated by PERK. STAT3 contributes to pro-inflammatory/immunosuppressive cytokine release and as we have recently shown, it up-regulates PD-L1 in EBV- infected monocytes, strongly impairing T cell function [24].
The same cytokines may activate also mTOR that is the master negative regulator of autophagy. Interestingly, autophagy is required for an efficient innate and specific immune response and its inhibition facilitates viral escape from immune recognition. Moreover, it may exacerbate inflammation, as autophagy is involved in the removal of apoptotic bodies and negatively regulates the activation of inflammosomes [25]. Inhibitors of STAT3 such as LLL-12 or SF-1066 employed for the treatment of CML patients or mTOR inhibitor such as Rapamycin or Metformin could be used to counteract autophagy inhibition by SARS-CoV-2 infected COVID19 patients and restore immunity. More importantly, as ER stress/UPR strongly regulates the production of inflammatory cytokines and ROS, we hypothesize that the reduction of ER stress and/or the manipulation of UPR arms may hold the key to tune inflammation, reducing its-induced tissue damage and counteract immune dysfunction in the course of infection by highly pathogenic viruses SARS-CoV2. Chemical chaperones such as 4-PBA or specific inhibitors of UPR arms such as IRE1α inhibitor 4m8c that inhibits its endonuclease activity or GSK2850163 that prevents also its kinase activity or PERK inhibitors such as GSK2606414 or ATF6 inhibitor Nelfinavir could be used at this purpose. UPR manipulation could be also accompanied by the use PRR agonists/antagonist that, as above said, may potentiate the anti-viral response and control the intensity of inflammation.
Conflict of Interest
I declare no conflict of interest.
Acknowledgment
The work Cirone’e laboratory was supported by AIRC IG 2019-23040 and by Istituto Pasteur Italia Fondazione Cenci Bolognetti.
References
-
Thompson MR, Kaminski JJ, Kurt Jones EA, Fitzgerald KA (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3(6): 920- 940.
-
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124(4): 783-801.
-
Jia HP, Look DC, Shi L, Melissa H, Lecia P, et al. (2005) ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol 79(23): 14614-14621.
-
Totura AL, Whitmore A, Agnihothram S, Alexandra S, Michael GK, et al. (2015) Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 6(3): e00638-e00615.
-
Spiegel M, Pichlmair A, Martinez Sobrido L, Jerome C, Adolfo García S, et al. (2005) Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol 79(4): 2079-2086.
-
Asha K, Sharma Walia N (2018) Virus and tumor microenvironment induced ER stress and unfolded protein response: from complexity to therapeutics. Oncotarget 9(61): 31920-31936. **7.** Cirone M (2020) Perturbation of bulk and selective macroautophagy, abnormal UPR activation and their interplay pave the way to immune dysfunction, cancerogenesis and neurodegeneration in ageing. Ageing Res Rev 58: 101026.
-
Fung TS, Liu DX (2014) Coronavirus infection, ER stress apoptosis and innate immunity. Front Microbiol 5: 296. **9.** Cirone M (2018) EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 10(11): 599.
-
Gassen NC, Niemeyer D, Muth D, Victor MC, Silvia M, et al. (2019)SKP2 attenuates autophagy through Beclin1- ubiquitination and its inhibition reduces MERS- Coronavirus infection. Nat Commun 10: 5770.
-
Bhattacharyya S, Sen U, Vrati S (2014) Regulated IRE1- dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J Gen Virol 95(1): 71-79.
-
Osorio F, Tavernier SJ, Hoffmann E, Yvan S, Liesbet M, et al. (2014) The unfolded-protein-response sensor IRE- 1alpha regulates the function of CD8alpha+ dendritic cells. Nat Immunol 15(3): 248-257.
-
Romeo MA, Gilardini Montani MS, Falcinelli L, Aurelia G, Cristina N, et al. (2019) HHV-6B reduces autophagy and induces ER stress in primary monocytes impairing their survival and differentiation into dendritic cells. Virus Res 273: 197757.
-
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, et al (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23(20): 7198-7209.
-
Reverendo M, Mendes A, Arguello RJ, Gatti E, Pierre P (2019) At the crossway of ER-stress and proinflammatory responses. FEBS J 286(2): 297-310.
-
Kim S, Joe Y, Surh YJ, Chung HT (2018) Differential Regulation of Toll-Like Receptor-Mediated Cytokine Production by Unfolded Protein Response. Oxid Med Cell Longev 24: 9827312.
-
Darling NJ, Cook SJ (2014) The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta 1843(10): 2150-2163.
-
Soto Pantoja DR, Wilson AS, Clear KY, Westwood B, Triozzi PL, et al. (2017) Unfolded protein response signaling impacts macrophage polarity to modulate breast cancer cell clearance and melanoma immune checkpoint therapy responsiveness. Oncotarget 8(46): 80545-80559.s
-
Cubillos Ruiz JR, Silberman PC, Rutkowski MR, Sahil C, Alfredo PP, et al. (2015) ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161(7): 1527-1538.
-
Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U (2005) Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res 96(12): 1233-1239.
-
Burman A, Tanjore H, Blackwell TS (2018) Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol 68- 69: 355-365.
-
Levi M, Keller TT, van Gorp E, Ten Cate H (2003) Infection and inflammation and the coagulation system. Cardiovasc Res 60(1): 26-39.
-
Sander WJ, O Neill HG, Pohl CH (2017) Prostaglandin E2 As a Modulator of Viral Infections. Front Physiol 8: 89.
-
Gilardini Montani MS, Santarelli R, Falcinelli L, et al. (2018) EBV up-regulates PD-L1 on the surface of primary monocytes by increasing ROS and activating TLR signaling and STAT3. J Leukoc Biol 104(4): 821-832.
-
Qian M, Fang X, Wang X (2017) Autophagy and inflammation. Clin Transl Med 6(1): 24.
- hMPV: Is It Another Covid-19 Like Situation?
- Streptomyces: Sources of Novel Discoveries in Antibiotic Research to Combat Antimicrobial Resistance
- A Review of Mosquitoes (Diptera: Culicidae) and Their Biodiversity, Medical and Veterinary Importance
- Past and Current Immunotherapy in Cancer
- Hematological Cancer and Viral Infection
- The Growing Threat of Antimicrobial Resistance in India: Challenges and Solutions