Computational Remodeling Mechanism Analysis of Three HIV Enzyme Partners (Protease (PR), Reverse Transcriptase (RT) and Integrase (IN)) on Behalf of Structural and Functional Analysis
Remodeling mechanism analysis of three HIV enzymes based on computational analysis for potential active sites identification of three viral enzymes, reverse transcriptase (RT), integrase (IN), and protease (PR) are all good drug targets and three distinct types of inhibitors, which block the polymerase activity of RT and IN, have been approved to treat HIV-1 infections, A better understanding of the structure and function of three HIV enzymes and mechanism of inhibition can be used to generate better drugs; in particular drugs that are effective against the current drug-resistant strains of HIV-1 and HIV 2 via structure prediction analysis of three HIV enzymes present their different activities as given tables. Computational protein modeling is a method designed for the most probable 3D structure from a sequence given its lignments with related structure .In this topic we discuss about the three HIV partner enzymes structure of HIV1and HIV2 from different software’s and their variations in core value, hydrophobicity, residues and intermolecular forces. Finally In this project we developed remodeled structures of different parameters mechanism in different core HIV enzymes partners with different types of functional structure prediction based on computational arithmetical calculations.
Introduction
AIDS is caused by HIV infection and is characterized by a severe reduction in CD4+ T cells, which means an infected person develops a very weak immune system and becomes vulnerable to contracting life-threatening infections (such as Pneumocystis carinii pneumonia). AIDS occurs late in HIV disease. Tracking of the disease in the United States began early after the discovery of the pandemic, but even to date, tracking data reveal only how many individuals have AIDS, not how many have HIV. The counted AIDS cases are like the visible part of an iceberg, while the much larger portion, HIV, is submerged out of sight. Many States are counting HIV cases now that positive results are to be gained by treating the infection in the early stages and because counting only AIDS cases is no longer sufficient for projecting trends of the pandemic. However, because HIV-infected people generally are asymptomatic for years, they might not be tested or included in the count. The CDC estimates that between 650,000 and 900,000 people in the United States currently are living with HIV.
In 1996, the number of new AIDS cases (not HIV cases) and deaths from AIDS began to decline in the United States for the first time since 1981. Deaths from AIDS have decreased since 1996 in all racial and ethnic groups and among both men and women. However, the most recent CDC data show that the decline is slowing. The decline can be attributed to advances in treating HIV with multiple medications, known as combination therapy; treatments to prevent secondary opportunistic infections; and a reduction in the HIV infection rate in the mid-1980s prior to the introduction of combination therapy. The latter can be attributed to improved services for people with HIV and access to health care. In general, those with the best access to good, ongoing HIV/AIDS care increase their chances of living longer [1].
Most antiretroviral drugs target three HIV-1 proteins: PR, RT, and IN. These proteins are highly variable: many different amino acids can be present at the same position in viruses from different individuals. Some of the amino acid variants cause drug resistance and occur mainly in individuals receiving antiretroviral drugs. Some variants result from a human cellular defense mechanism called APOBEC-mediated hyper mutation. Many variants result from naturally occurring mutation. Some variants may represent technical artifacts. We studied PR and RT sequences from >100,000 individuals and IN sequences from >10,000 individuals to quantify variation at each amino acid position in these three HIV-1 proteins. We performed analyses to determine which amino acid variants resulted from antiretroviral drug selection pressure, APOBEC-mediated editing, and naturally occurring variation. Our results provide information essential to clinical, research, and public health laboratories performing genotypic resistance testing by sequencing HIV- 1 PR, RT, and IN [2].
Integrase is a 288-amino acid protein (32 kDa) encoded by the end of the pol gene. It is produced as part of the Gag-Pol polypeptide precursor, from which it is released by viral protease-mediated cleavage. It has three independent domains: (i) The N-terminal domain (amino acids 1-49) that carries an HHCC motif analogous to a zinc finger, and effectively binds Zn2+ possibly favoring protein multimerisation, a key process in integration [3, 4, 5]. (ii) The central domain or catalytic domain (amino acids 50-212) encompassing a D, D-35, E motif which is indispensable for the catalytic activity and which is conserved between viral IN and transposes. This central domain is also implicated in the binding of the viral DNA extremities mainly via the residues Q148, K156 and K159 [6, 7]. All integrase activities strictly require the presence of a metallic cationic cofactor which is coordinated by two residues of the catalytic triad (D64 and D116 for HIV-1 IN) (iii) The C-terminal domain (amino acids 213-288) binds non-specifically to DNA and therefore is mainly involved in the stability of the complex with DNA. No complete structure has yet been determined for the integrase protomer (IN1-288), or for oligomers or complexes of these structures with DNA, due to poor solubility and inters domain flexibility problems. However, several structures of isolated domains or of two consecutive domains have been reported [8, 9, 10, 11, 12, 13, 14, 15, 16].
PR belongs to the family of aspartic proteases. The structure of PR is a homodyne and consists of subunits of 99 amino acid residues each subunit is made up of nine β-strands and a single α-helix. Four anti-parallel β-strands form the highly stable dimer interface which constitutes the active site the core of the active site is hydrophobic and contains two aspartic acid residues contributed by both subunits. Flexible anti-parallel β-sheets from both monomers form two flaps that cover the active site thereby restricting access to it in the free enzyme state, the flaps assume a semi-open conformation and with a ligand in the active site, they assume a closed conformation it has been reported that, a network of weakly polar interactions between the flaps keeps them in a semi-open conformation Two models have been used to explain the mechanisms of flap opening and closing. In the first model, the ligand forms a collision complex with HIV-1 PR in the open flap conformation as it enters the active site of the PR and then induces the flaps to close in the second model, the ligand approaches the HIV-1 PR in the semi-open flap conformation and then induces the flaps to adopt an open conformation as it enters the active site [17, 18, 19].
Distinct HIV subtypes, circulating recombinant forms (CRF) and quasispecies have been isolated from infected patients due to the high degree of HIV genetic variability a consequence of the selective pressure of the host immune system and/or antiretroviral therapy. When genetic variability is present in HIV, protease (PR) and/or reverse transcriptase (RT) genes are particularly important. Drug classes against these targets are still used as first-line antiretroviral treatment. The PR and RT enzyme variability can decrease HIV fitness and leads to further variability as a compensatory mechanism for virus propagation [20, 21].
Material and Methodology
The genome sequences of the species were retrieved from National Centre for Biological Information (NCBI) (www.ncbi.nlm.nihgov). Statistical analysis was done via Geno 3D server then functional study done via mVISTA and Red star tool-UNITED KINGDOM and other online available tools for this computational study.
Results and Discussion
Computational remodeling mechanism analysis of three HIV enzyme partners study done on behalf of 10 models prediction based on three partner enzymes: Structural & Functional. Remodeling study of three HIV partners’ enzymes was done via online available Tools and Statistical analysis on behalf of different parameters based on Geno3D Predictions. After all predictions comparison we found so many differences of the enzymes via all these prediction we found if we can control all malfunctions of the enzymes on primitive level when enzyme correlate with receptor on first phase so we can control the mal function of the enzymes based on target of the drugs (Figure 1).
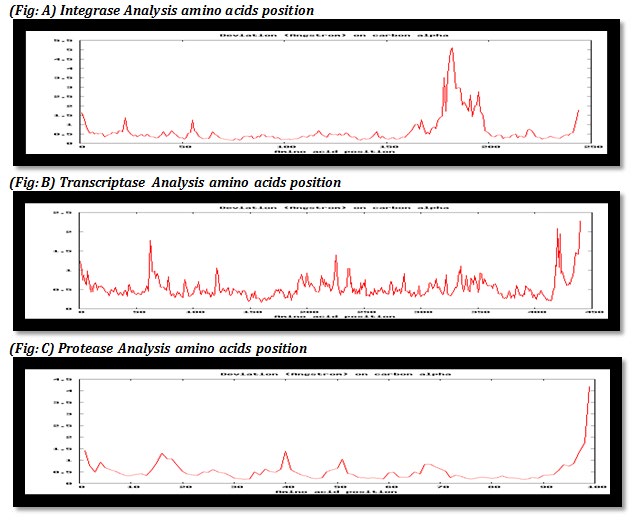
Figure 1 (A, B, C): In above table graph explain mean deviation of all the 10 models on carbon alpha in amino acids of three enzymes integrase, protease and transcriptase based on free amino acids positions.
This table explains arithmetical calculations of three enzymes partners with arithmetical means values of core region and frees residual allowed and disallowed molecules (Table 1).
| Integrated Analysis | |||||
|---|---|---|---|---|---|
| Models | Models Energy (Kcal/mol) | Core | Allowed | Generously | Disallowed |
| Model 1 | -10783 | 75.00% | 22.60% | 1.40% | 0.90% |
| Model 2 | -10803.1 | 71.20% | 25.90% | 0.50% | 2.40% |
| Model 3 | -10665.1 | 71.20% | 23.60% | 2.40% | 2.80% |
| Model 4 | -10777.7 | 75.00% | 21.70% | 1.40% | 1.90% |
| Model 5 | 10789.9 | 74.50% | 22.60% | 0.90% | 1.90% |
| Model 6 | -10611.1 | 73.60% | 22.60% | 2.80% | 0.90% |
| Model 7 | -10676 | 76.90% | 19.80% | 1.90% | 1.40% |
| Model 8 | -10515.4 | 77.40% | 19.80% | 2.40% | 0.50% |
| Model 9 | -10745.9 | 75.00% | 21.20% | 2.80% | 0.90% |
| Model 10 | -10858.4 | 76.90% | 18.90% | 2.40% | 1.90% |
| Protease Analysis | |||||
| Models | Models Energy (Kcal/mol) | Core | Allowed | Generously | Disallowed |
| Model 1 | -4059.95 | 85.00% | 13.80% | 1.30% | 0.00% |
| Model 2 | -3921.1 | 92.50% | 7.50% | 0.00% | 0.00% |
| Model 3 | -4076.7 | 78.80% | 21.30% | 0.00% | 0.00% |
| Model 4 | -4044.68 | 80.00% | 20.00% | 0.00% | 0.00% |
| Model 5 | -3926.24 | 88.80% | 11.30% | 0.00% | 0.00% |
| Model 6 | -4126.18 | 83.80% | 16.30% | 0.00% | 0.00% |
| Model 7 | -4072.28 | 80.00% | 18.80% | 1.30% | 0.00% |
| Model 8 | -3930.51 | 88.80% | 10.00% | 1.30% | 0.00% |
| Model 9 | -4090.83 | 87.50% | 12.50% | 0.00% | 0.00% |
| Model 10 | -4053.38 | 80.00% | 20.00% | 0.00% | 0.00% |
| Reverse Transcriptase Analysis | |||||
| Models | Models Energy (Kcal/mol) | Core | Allowed | Generously | Disallowed |
| Model 1 | -20021.1 | 80.80% | 17.40% | 1.30% | 0.50% |
| Model 2 | -19795.7 | 78.20% | 17.40% | 2.40% | 2.10% |
| Model 3 | -19877.4 | 80.50% | 17.40% | 0.50% | 1.60% |
| Model 4 | -19692 | 78.20% | 18.20% | 2.10% | 1.60% |
| Model 5 | -19924.6 | 79.20% | 18.90% | 0.80% | 1.10% |
| Model 6 | -19994.4 | 80.30% | 17.40% | 1.80% | 0.50% |
| Model 7 | -19871 | 79.70% | 17.40% | 2.10% | 0.80% |
| Model 8 | -19615.9 | 77.60% | 19.70% | 1.60% | 1.10% |
| Model 9 | -19667.8 | 77.10% | 19.50% | 2.10% | 1.30% |
| Model 10 | -19964 | 80.80% | 16.80% | 1.60% | 0.80% |
Table: 1 Arithmetical calculation of all three partners’ enzymes.
This table explain Ramachandran plot in which red area represents core with most favorable values of torsion angles and in which ideally 90% of residue in core region are present, column 2 explains ramachandran plots for all the amino acids with no of residues shown in brackets column 3 explains comparison with well refined structures, column 4 explains standard deviation according to torsion angles and column 5 explains maximum deviation ,absolute torsion and g factor for all the residues (Table 2).
This table explain Ramachandran plot in which red area represents core with most favorable values of torsion angles and in which ideally 90% of residue in core region are present, column 2 explains ramachandran plots for all the amino acids with no of residues shown in brackets column 3 explains comparison with well refined structures, column 4 explains standard deviation according to torsion angles and column 5 explains maximum deviation ,absolute torsion and g factor for all the residues (Table 3).
This table explain Ramachandran plot in which redred area represents core with most favorable values of torsion angles and in which ideally 90% of residue in core region are present, coloum 2 explains ramachandran plots for all the aminoacids with no of residues shown in brackets coloum 3 explains comparison with well refined structures, coloum 4 explains standard deviation according to torsion angles and coloum 5 explains maximum deviation ,absolute torsion and g factor for all the residues with statically and structural differences (Table 4).
| Integrase Analysis | |||||
|---|---|---|---|---|---|
| Models | Ramchandran Plot | Ramchandran Plot for all residue | Main Chain Parameters | Side chain parameters | Residue Properties |
| Model 1 | |||||
| Model 2 | |||||
| Model 3 | |||||
| Model 4 | |||||
| Model 5 | |||||
| Model 6 | |||||
| Model 7 | |||||
| Model 8 |
Table 2: Analysis of Reverse transcriptase with all residual structures.
| Model 9 | |||||
|---|---|---|---|---|---|
| Model 10 |
Table 3: Analysis of Reverse transcriptase with all residual structures.
Table: 2 Analysis of integrase with all residual structures.
| Protease Analysis | |||||
|---|---|---|---|---|---|
| Models | Ramchandran plot | Ramchandran plot for all residue | Main chain parameters | Side chain parameters | Residue types |
| Model 1 | |||||
| Model 2 |
Table 4: Analysis of Reverse transcriptase with all residual structures.
| Model 3 | |||||
|---|---|---|---|---|---|
| Model 4 | |||||
| Model 5 | |||||
| Model 6 | |||||
| Model 7 |
Table 5: Analysis of Reverse transcriptase with all residual structures.
| Model 8 | |||||
|---|---|---|---|---|---|
| Model 9 | |||||
| Model 10 | |||||
| Reverse transcriptase | |||||
| Models | Ramchandran plot | Ramchandran plot for all residue | Main chain parameters | Side chain parameters | Residue types |
| Model 1 | |||||
| Model 2 |
Table 6: Analysis of Reverse transcriptase with all residual structures.
| Model 3 | |||||
|---|---|---|---|---|---|
| Model 4 | |||||
| Model 5 | |||||
| Model 6 | |||||
| Model 7 | |||||
| Model 8 | |||||
| Model 9 | |||||
| Model 10 |
Table 7: Analysis of Reverse transcriptase with all residual structures.
Conclusion
Remodeling of all the three main enzymes integrase, protease and transcriptase with help of Geno 3D server to obtain potentially active sites of three viral enzymes which can be targeted by drugs in treatment of HIV patients, detailed analysis in the form of models energy, Ramachandran plot and its parameters and correlation between them can be used in successful drug designing.
References
-
https://www.ncbi.nlm.nih.gov/books/NBK64928/
-
Rhee SY, Sankaran K, Varghese V, Win- ters MA, Hurt CB, et al. (2016) HIV 1 Protease, Re- verse Transcriptase, and Integrase Variation. ASM 90: 1-13.
-
Zheng R, Jenkins TM, Craigie R (1996) Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc Natl Acad Sci USA 93(24): 13659-13664.
-
Lee SP, Xiao J, Knutson JR, Lewis MS, Han MK (1997) Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36: 173-180.
-
Esposito D, Craigie R (1998) Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J 17(19): 5832-5843.
-
Jenkins TM, Esposito D, Engelman A, Craigie R (1997) Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo- crosslinking. EMBO J 16(22): 6849-6859.
-
Heuer TS, Brown PO (1997) Mapping features of HIV- 1 integrase near selected sites on viral and target DNA molecules in an active enzyme-DNA complex by photo- cross-linking. Biochemistry 36(35): 10655-10665.
-
Drake RR, Neamati N, Hong H, Pilon AA, Sunthankar P, et al. (1998) Identification of a nucleotide binding site in HIV-1 integrase. Proc Natl Acad Sci USA 95(8): 4170- 4175.
-
Johnson AA, Santos W, Pais GC, Marchand C, Amin R, et al. (2006) Integration requires a specific interaction of the donor DNA terminal 5’-cytosine with glutamine 148 of the HIV-1 integrase flexible loop. J Biol Chem 281(1): 461-467.
-
Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, et al. (1998) Three new structures of the core domain of HIV- 1 integrase: an active site that binds magnesium. Proc Natl Acad Sci USA 95(16): 9150-9154.
-
Maignan S, Guilloteau JP, Zhou Liu Q, Clement Mella C, Mikol V (1998) Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J Mol Biol 282(2): 359-368.
-
Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, et al. (1997) Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat Struct Biol 4(7): 567-577.
-
Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, et al. (1995) Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 34: 9826-9833.
-
Wang JY, Ling H, Yang W, Craigie R (2001) Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J 20(24): 7333-7343.
-
Chen JC, Krucinski J, Miercke LJ, Moore FJS, Tang AH (2000) Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA 97(15): 8233-8238.
-
Imamichi T (2004) Action of anti-HIV drugs and resistance: reverse transcriptase inhibitors and protease inhibitors. Curr Pharm Des 10(32): 4039-4053.
-
Dayam R, Neamati N (2003) Small-molecule HIV-1 integrase inhibitors: the 2001-2002 update. Curr Pharm Des 9(22): 1789-1802.
-
Neamati N, Marchand C, Pommier Y (2000) HIV-1 integrase inhibitors: past, present, and future. Adv Pharmacol 49: 147-165.
-
Pommier Y, Marchand C, Neamati N (2000) Retroviral integrase inhibitors year 2000: update and perspectives. Antiviral Res 47(3): 139-148.
-
Brik A, Wong CH (2003) HIV-1 protease: mechanism and drug discovery. Org Biomol Chem 1(1): 5-14.
-
Hoggard PG, Owen A (2003) The mechanisms that control intracellular penetration of the HIV protease inhibitors. J Antimicrob Chemother 51(3): 493-496.
- hMPV: Is It Another Covid-19 Like Situation?
- Streptomyces: Sources of Novel Discoveries in Antibiotic Research to Combat Antimicrobial Resistance
- A Review of Mosquitoes (Diptera: Culicidae) and Their Biodiversity, Medical and Veterinary Importance
- Past and Current Immunotherapy in Cancer
- Hematological Cancer and Viral Infection
- The Growing Threat of Antimicrobial Resistance in India: Challenges and Solutions