The Molecular Docking and Dynamic Analyses of 3',5-Dihydroxy- 4',6,7-Trimethoxyflavanone, Isovitexin-2'-O-Rhamnoside, Solafuranone, (R)-5,3'-Dimethyl hesperidin, Phloretin 3',5'-Di-C -glucoside, Isomargaritene, Margaritene and Clemomandshuricoside B for Potential COVID-19 Therapy
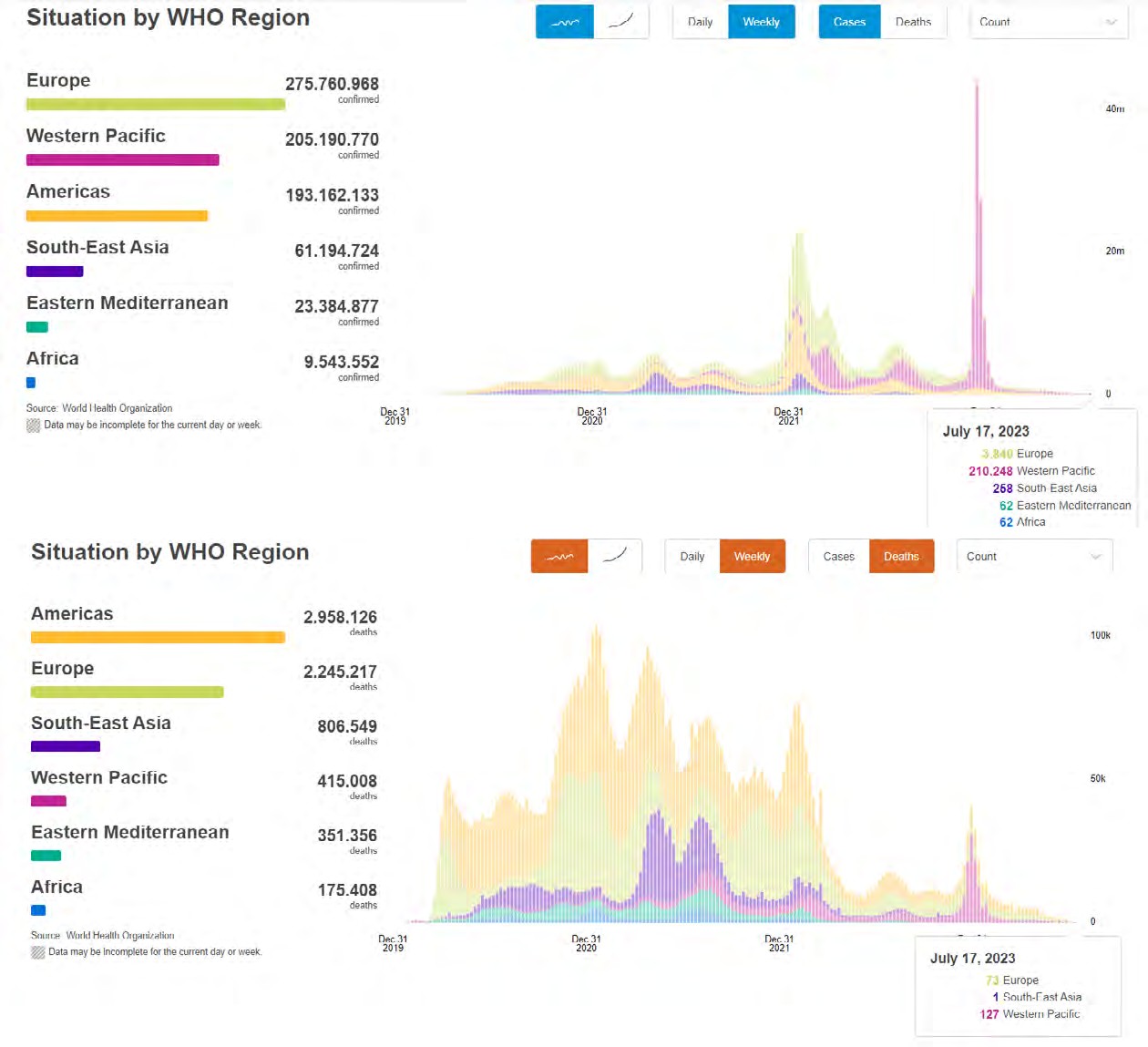
Covid-19 (SARS-CoV-2), a new type of coronavirus, first appeared in the Chinese city of Wuhan in December 2019 and has become the main cause of the pandemic today. Although molecular studies continue, vaccination studies are not sufficient. In this Covid-19 process, which provides further increase in antiviral drug discoveries, we perform ligand-protein interactions in order to find an antiviral active substance with this study, Autodock. We present the analyzes based on the comparison of 8 different ligands(3',5-Dihydroxy-4',6,7-trimethoxyflavanone, Isovitexin-2'-O-rhamnoside, Solafuranone, (R)-5,3'-Dimethyl hesperidin, Phloretin 3',5'-Di-C-glucoside, Isomargaritene, Margaritene and Clemomandshuricoside B) based on the 6LU7 protein. It was observed that the molecules were bound by interacting with the main protease of Covid-19. Thus, we aim to be a reference with you in this article for further studies that enable ligands to create drug potential. Our results will serve as a reference for this molecular docking study.
Introduction
The coronavirus responsible for the 2019 pandemic, officially named SARS-CoV-2, originated in Wuhan, China’s Hubei Province, and has since spread globally, causing a significant impact on public health and the world economy.
Coronaviruses are a family of viruses known for their crown-like spikes on their outer surfaces, from which they got their name (“corona” means crown in Latin). They are classified within the family Coronaviridae and belong to the order Nidovirales. Viruses in this family have a nucleic acid genome, which can be RNA or DNA. Coronaviruses, including SARS-CoV-2, are single-stranded RNA viruses [1].
The genome size of coronaviruses can vary, but they usually have nucleic acid genomes ranging in length from 26 to 32 kilobases. Despite their relatively large genome size, coronavirus particles are quite small and typically have diameters ranging from 65 to 125 nanometers [1, 2].
Coronaviruses are further divided into four different subgroups: alpha, beta, gamma, and delta. SARS-CoV-2 belongs to the beta-coronavirus subgroup. Other well- known coronaviruses such as SARS-CoV (responsible for the Severe Acute Respiratory Syndrome outbreak 2002-2003) and MERS-CoV (responsible for the Middle East Respiratory Syndrome outbreak 2012) are also beta-coronaviruses.
The genetic diversity and classification of coronaviruses is crucial to understanding their epidemiology, pathogenesis, and potential to cause disease outbreaks. As the ongoing pandemic has shown, coronaviruses, especially those in the beta subgroup, can pose significant public health problems and require extensive research and global collaboration for containment and mitigation efforts [1, 2, 3].
Various coronaviruses that cause respiratory illness can be transmitted to humans. SARS-CoV-2 is a virus that stands out with its higher mortality rate than other coronaviruses and its ability to cause acute respiratory syndrome. Since SARS-CoV-2 is a highly pathogenic coronavirus strain, the second group of the Beta-coronavirus genus is included [2, 3].
SARS-CoV-2 is an enveloped virus with positive single- stranded RNA. This virus has also been detected in bats and has 96.2% genetic similarity to a virus isolated from bats called CoV RaTG13. It also shows 88% identity with the bat SL-CoVZC45 and bat SL-CoVZXC21 sequences [4].
The most distinctive feature of the coronavirus shows that the virus can be transmitted to humans from different animal species. These animals include hosts such as snakes, turtles and pangolins. These animals have the potential to transmit the virus from Malaysia to China [5].
In particular, transmission of SARS-CoV-2 to humans occurs through airborne droplets and contact. Therefore, it is important to take precautions such as hygiene measures, wearing masks, social distancing and vaccination to prevent transmission. Research continues to understand user origin and infection groups [6, 7].
In silico studies include computational methods and simulations to analyze biological and chemical interactions, and these have become common in various scientific fields, including medicine, molecular biology, pharmacology, and toxicology.
In the context of SARS-CoV-2, researchers have found sequence similarities between its protein-coding gene structures and those of other coronaviruses such as MERS- CoV and SARS-CoV [8]. While the similarity with MERS-CoV is 51%, the similarity with SARS-CoV is 79.5%. SARS-CoV-2, similar to SARS-CoV, uses the ACE2 receptor in the human body for its functioning. Understanding these similarities and receptor interactions is crucial for developing inhibitor studies and potential treatments for Covid-19 [9].
In silico research uses molecular docking and dynamics to analyze how potential drug candidates interact with target molecules and receptors. Computer-aided software allows researchers to calculate the proximity of receptors and target structures and test compounds with physics-based equations [10].
Plants contain bioactive compounds with various chemical properties, such as terpenes, alkaloids, phenolics and flavonoids, which display various biological properties. Throughout history, these compounds have been used in the treatment of various diseases. In the aforementioned study, the potential of specific bioactive compounds to be used as drugs in the treatment of Covid-19 was analyzed by in silico docking analysis, molecular dynamics, pharmacokinetic analysis, and binding free energy calculation [11, 12].
In silico studies provide valuable information and can serve as a starting point for further in vivo and in vitro studies. However, it is important that findings from in silico analyzes be confirmed by rigorous laboratory and clinical trials before evaluating any bioactive compound as a potential drug for the treatment of Covid-19. The drug development process is complex and requires careful evaluation and testing to ensure safety and efficacy [13, 14].
Material and Methods
Molecular Docking Analyzes
Calculations required for molecular docking were performed with Autodock Vina software [15]. Water molecules and cofactor structures were extracted from the protein via software to view and analyze the linkage between protein and ligand in detail [16]. The main protease of the virus belonging to Covid-19 and used as a protein was retrieved from the RSCB Protein Data Bank as a three- dimensional theoretical model (PDB ID: 6LU7). Accordingly, eight ligands were tested. Tested ligands and their properties are presented in Table 2 (https://pubchem.ncbi.nlm.nih. gov/). The binding potentials of hydroxychloroquine and chloroquine were reviewed as control ligands and used in the software. The two-dimensional form of the ligand structures was transformed into an energy-minimized three-dimensional conformation. All proteins and ligands used in the study were confirmed before performing in silico calculations [17].
Molecular Dynamic Analyzes
The ligand-protein complex was simulated using the WebGro tool [18]. To assess the stability of the complex, molecular dynamics (MD) simulation was conducted over a duration of 50 nanoseconds [19, 20, 21, 22, 23]. We optimized the MM/PB(GB)SA performance for accurate ligand binding conformations, utilizing both the Schrödinger package and the Amber package [24].
Drug Similarity and ADMET Prediction for Potential Covid-19 Inhibitor
Today, computer-based ADME analyzes have created an important study potential for drug discovery. The drug similarity, pharmacodynamics, and pharmacokinetics of the drug candidate molecule or molecules were performed with Swiss ADME, an online software (http://www.sib.swiss) (http://www.swissadme.ch/index. php) [25, 26]. In addition, these toxicological and pathological estimates were also applied to the Ghose, Veber, and Lipinski subjects and their bioavailability scores [27, 28].
Results and Discussion
For SARS-CoV-2, AutoDock Vina was screened with protein ligands and Covid markers to screen for potential drugs through a molecular insertion containing structural and non-structural protein regions. The pharmacokinetic properties of 8 ligands bound to 6LU7 of SARS-CoV-2 were determined. It has been observed that the molecular insertion results obtained with this study show a good interaction with the COVID-19 protease as potential drug candidate molecules. Function identification was performed using a scoring based on the Lamarckian General Algorithm of attachment strength. In the study, a molecular insertion based on antiviral mechanism was in question, as the flavonoid, Sesquiterpenes, Phenylpropanoids provide high affinity in the same regions of the main protease of SARS- CoV-2 as the control molecule. Bonding free energy is found in hydrogen bonding, van der Waals forces, and electrostatic identification. Identifying the lowest binding energy signifies the most stable interaction between the ligand and protein [29]. The computed binding energy outcomes usingAutodock vina are presented in Table 1. After positioning, all structures were visualized in VMD. It has comparable binding affinity (-5.9 to -11.7 kcal/mol) for all ligands, respectively (-11.7 and -11 kcal/mol). RSMD is a very important parameter used to analyze the stability of MD trajectories. Estimation of backbone atoms of protein and ligand-protein complexes is obtained by RSMD. Backbone RMSD measurements allowed us to gain insight into the conformational stability for the complexes. The RMSF of the backbone atoms of each residue in the 6LU7-linked ligand-protein complex was analyzed to gain insight into the flexibility of the enzyme backbone structure. A high RMSF value indicates more flexibility, while a low RMSF value indicates limited movements.
According to the analysis of molecular dynamics results, the residual number C mean square deviation (RMSD), root mean square fluctuation (RMSF), MD, which is a function of the simulation time, were used in the simulation to analyze the stability of the modeled areas.
| H bound | Energy | |
|---|---|---|
| 3’,5-Dihydroxy-4’,6,7-trimethoxyflavanone | 4 (Leu141;Ser144;His163;Gly143) | -8.5 kcal/mol |
| Isovitexin-2’’-O-rhamnoside (2’’-O-alpha-L- Rhamnopyranosyl-isovitexin) | 5(Thr199(2);Asn238;Leu287;Arg131) | -11.7 kcal/mol |
| Solafuranone | 0 | -5.9 kcal/mol |
| (R)-5,3’-Dimethyl hesperidin | 8 (Tyr239(2);Leu287;Lys137;Arg131;Asn238;Asp197(2) | -9.7 kcal/mol |
| Phloretin 3’,5’-Di-C-glucoside | 4 (Asp197; Asp289;Leu287; Tyr 239) | -10.3 kcal/mol |
| Isomargaritene | 8 (Met276;Tyr239(2);Thr199;Asp289;Asp197;Tyr237(2) | -11 kcal/mol |
| Margaritene | 3 (Glu166;Gln189;Leu141) | -9.9 kcal/mol |
| Clemomandshuricoside B | 5 (Lys5; Gln127; Lys137; Glu290; Glu288) | -10.1 kcal/mol |
Table 1: Results of the molecular docking between the target protein and the candidate drug molecules (ligands).
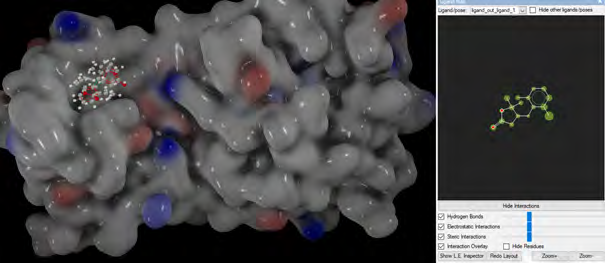

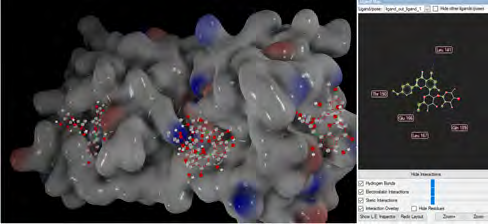
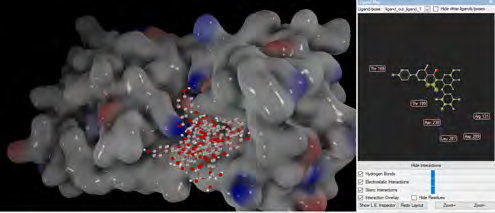
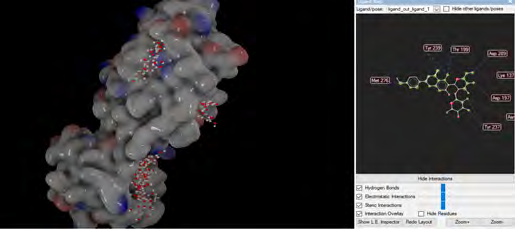
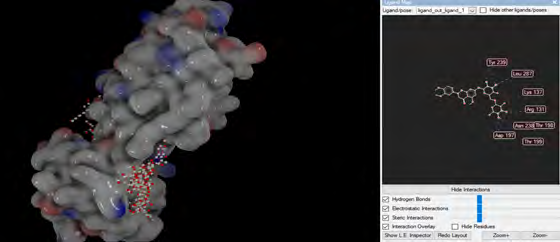
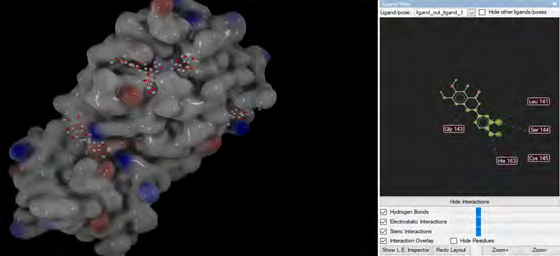
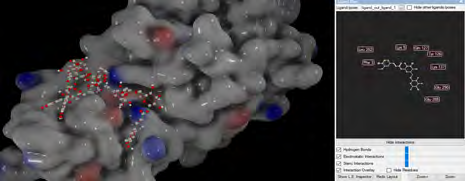
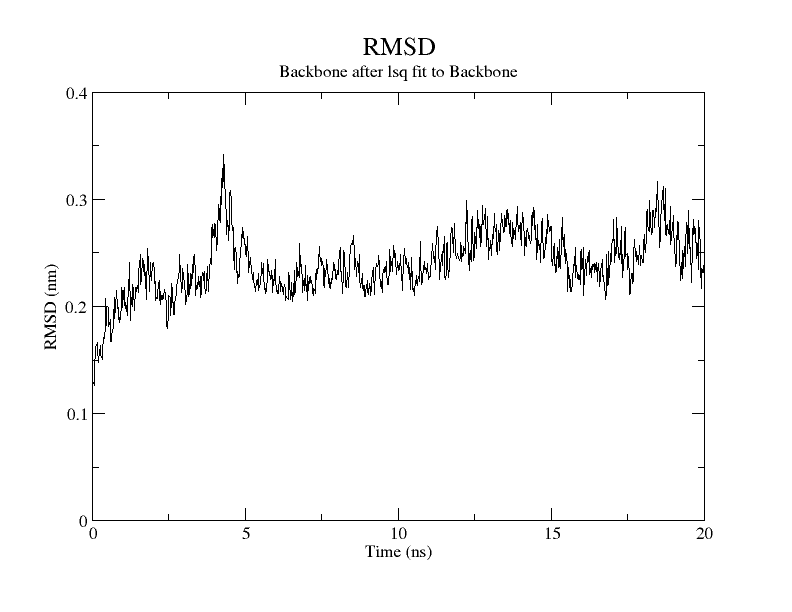
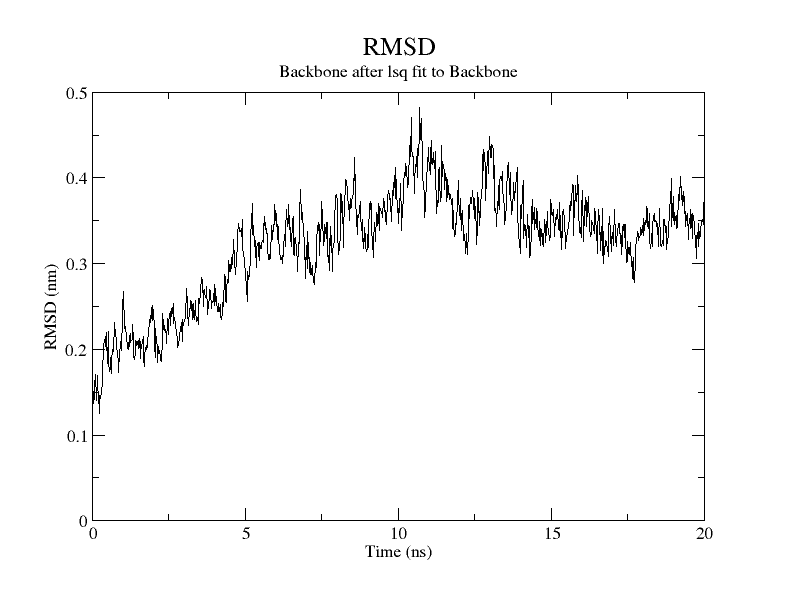
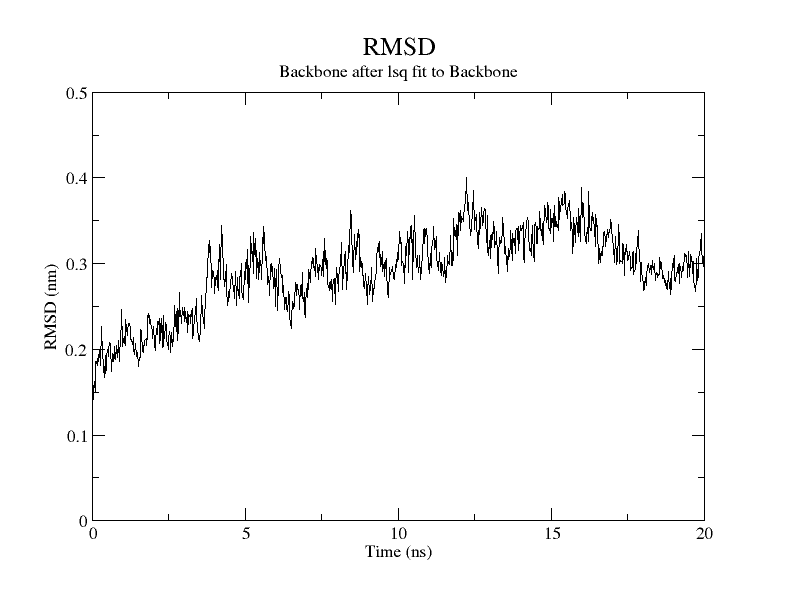
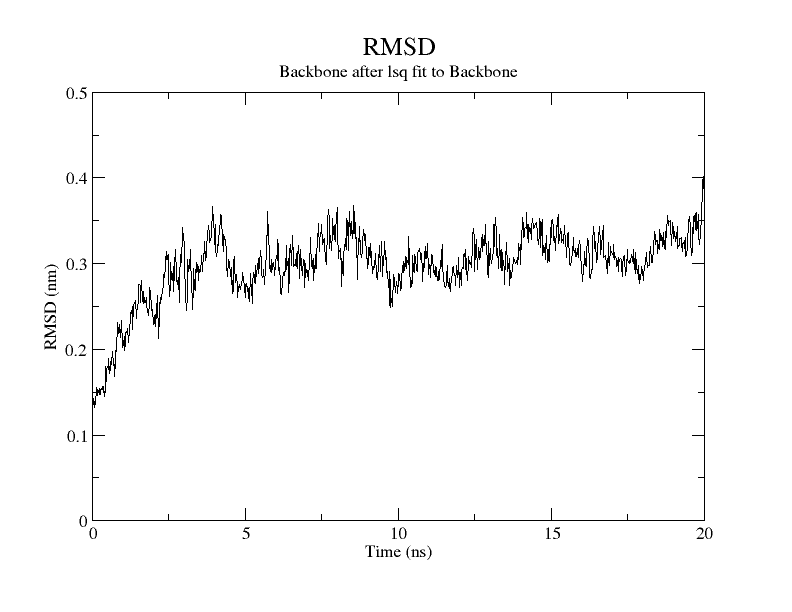
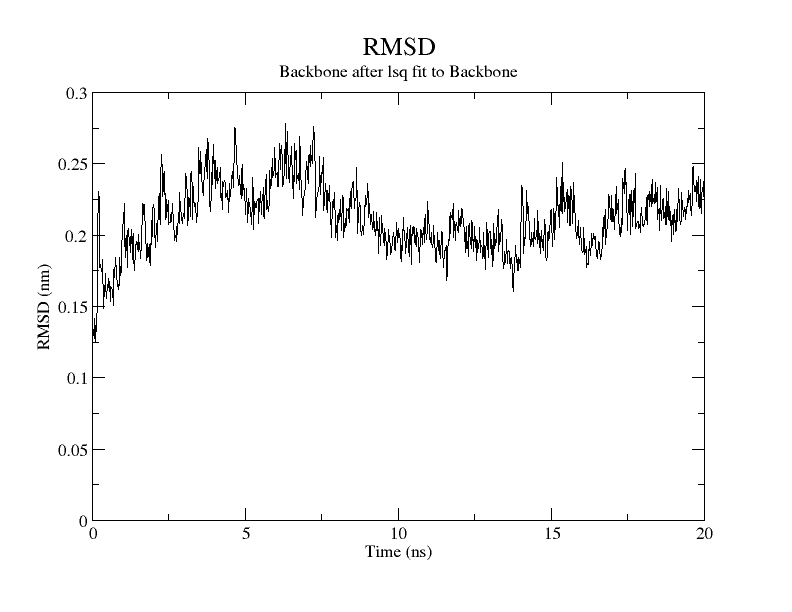
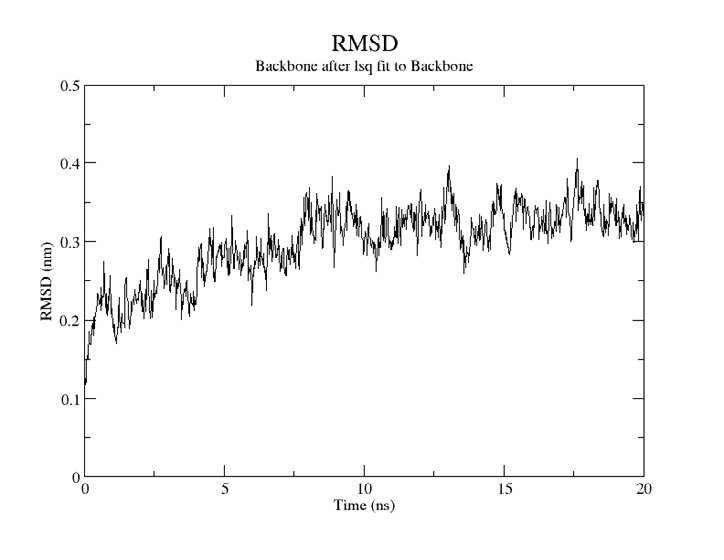
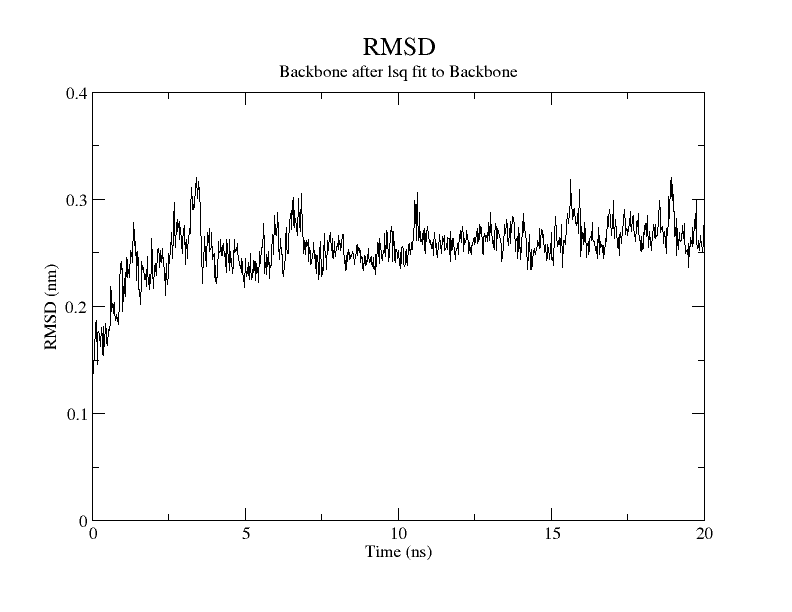
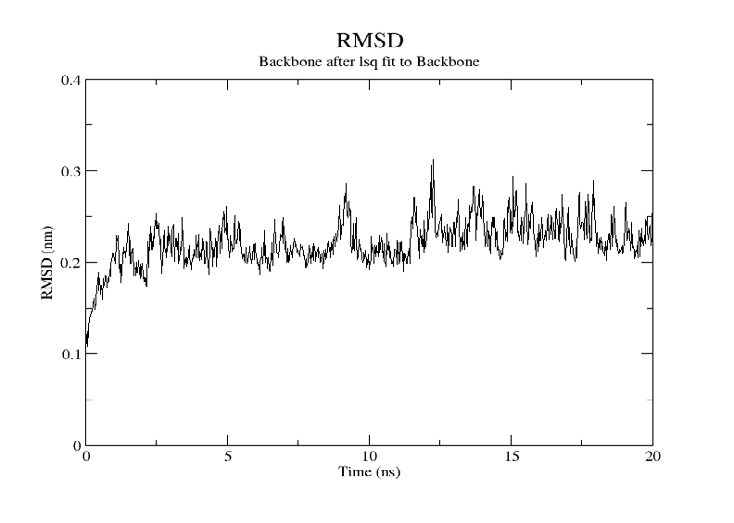
As performing molecular insertion scoring did not reveal an available estimate for ligand binding affinities, MM/ PB(GB)SA analyzes were performed to predictively analyze binding affinities. The binding free energy of MM/PB(GB)SA on behalf of ligands, including the Amber and Schrödinger package (http://cadd.zju.edu.cn/farppi) was defined [30].
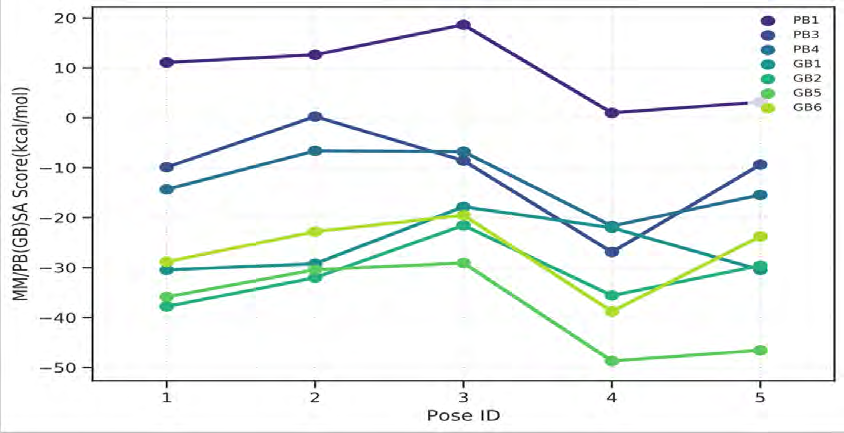
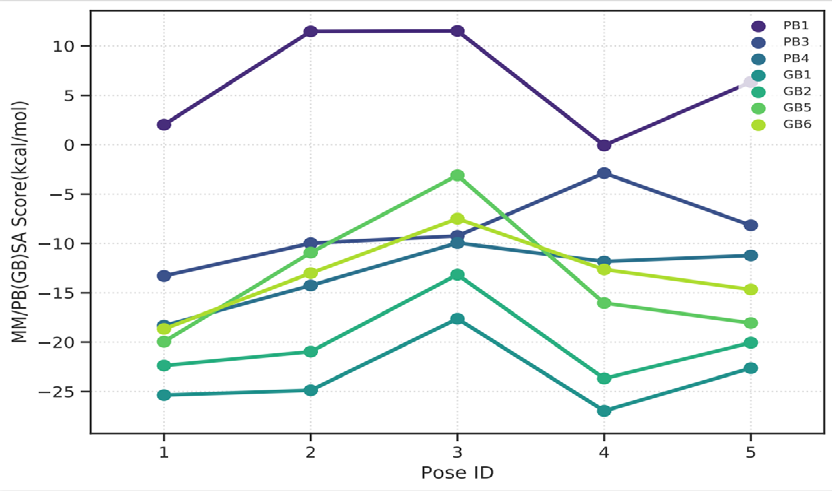
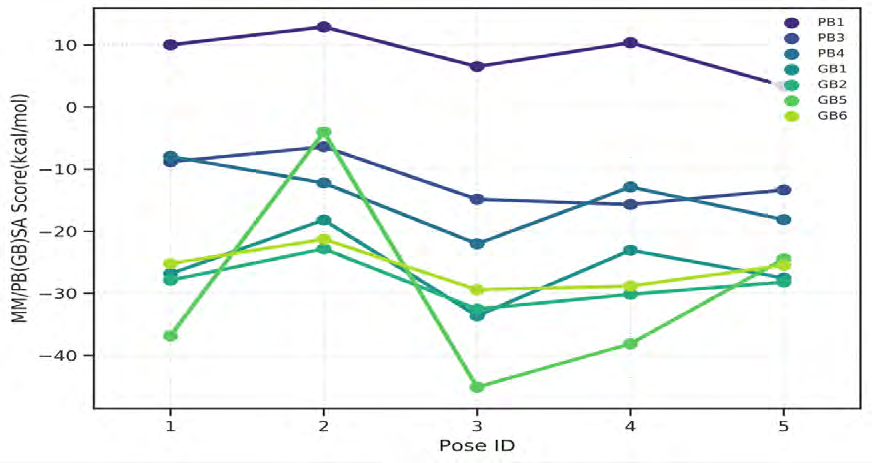
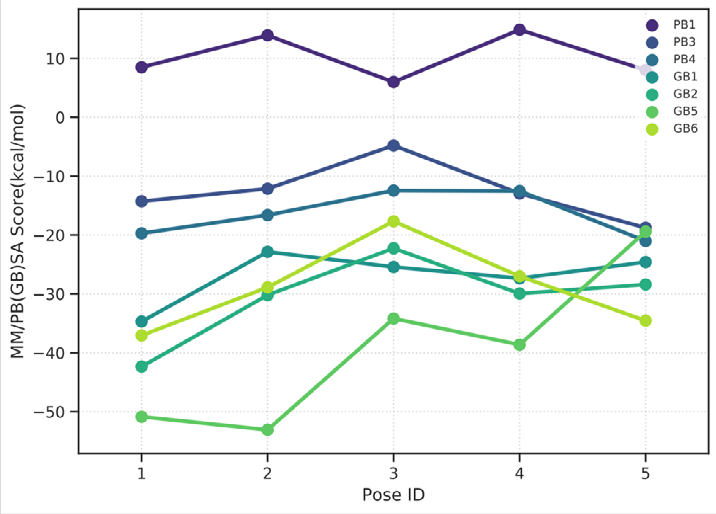
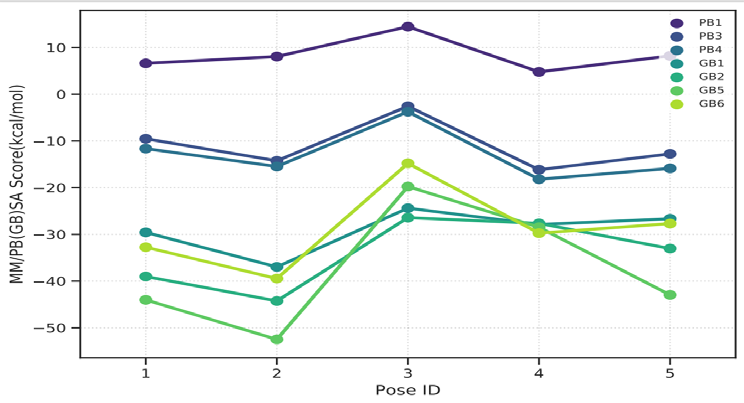
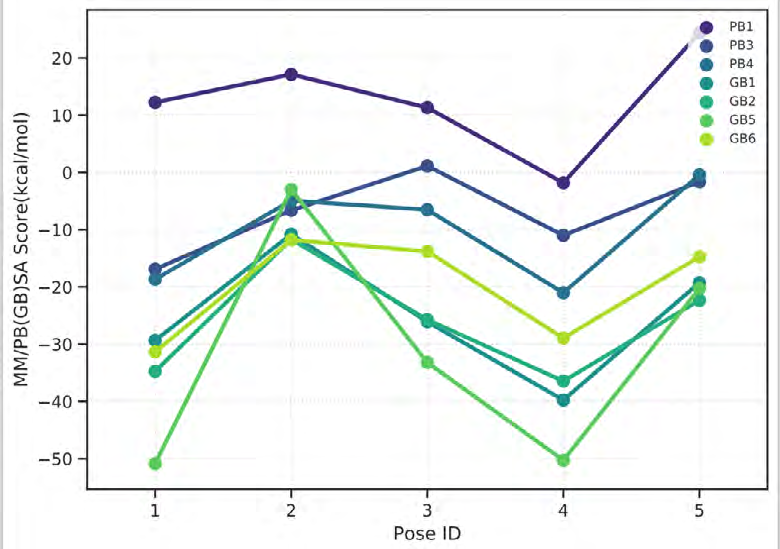
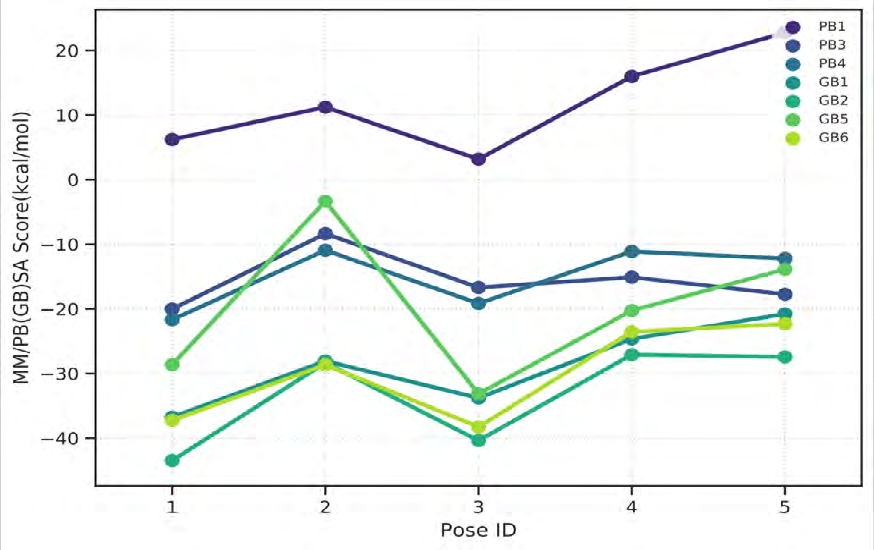
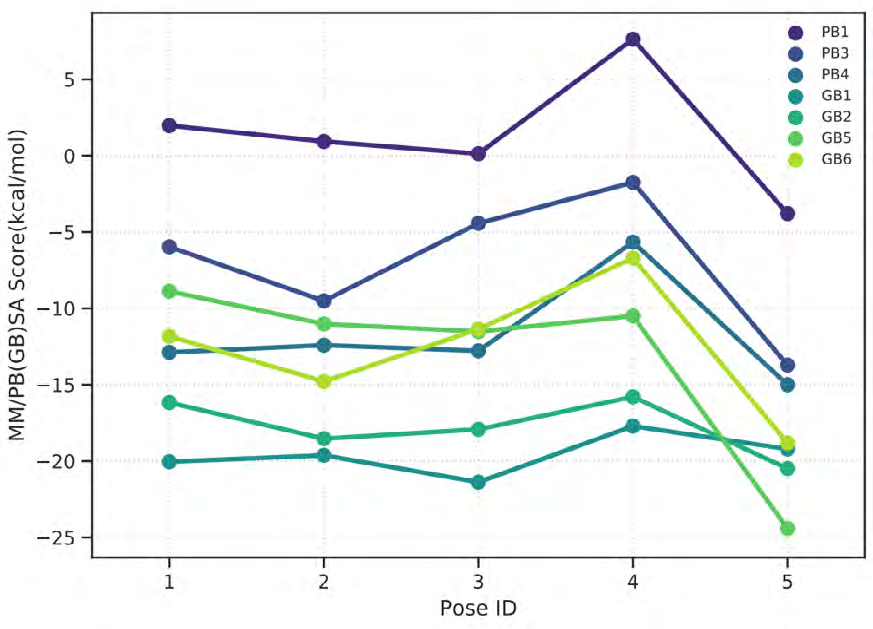
ADME properties of ligand structures were analysed ın SWISS-ADME and results were shown in Figure 26-33 (http://www.swissadme.ch/). In addition to drug similarity results, depending on Lipinski’s five rules (RO5, Pfizer rule), molecular weight (MW)<500, LogP≤5, hydrogen bond acceptors ≤10, including the violation of at most one of the following features [31]. For the active drug, the hydrogen bond total should tend to be ≤12, the rotatable bonds ≤10, and the polar surface area ≤140 oral, and ≥20 for bioavailability, given Veber’s rules. In short, only half of all FDA-approved small molecules comply with both the rule of five. Based on this, these analyzes reflect the potential to be used as a drug in drug development studies that are currently leading the way [32]. According to the results obtained, Solafuranone and 3’,5-Dihydroxy-4’,6,7-trimethoxyflavanone molecules have the potential to be drugs according to the Lipinski Ghose and Veber rules.
| Molecules | Plant | Phytochemicals | 3D structure |
|---|---|---|---|
| 3',5-Dihydroxy-4',6,7-trimethoxyflavanone | Salvia plebeia | Flavanoid | |
| Isovitexin-2'-O-rhamnoside (2'-O-alpha-L-Rhamnopyranosyl-isovitexin) | Crataegus pinnatifida Bunge | Flavanoid | |
| Solafuranone | Solanum indicum | Sesquiterpenes | |
| (R)-5,3'-Dimethyl hesperidin | Citrus sinensis | Flavanoid | |
| Phloretin 3',5'-Di-C-glucoside | Nothofagus fusca | Flavanoid | |
| Isomargaritene | Vitex trifolia L., | Flavanoid | |
| Margaritene | Vitex trifolia L., | Flavanoid | |
| Clemomandshuricoside B | Catalpa fargesii f. duclouxii | Phenylpropanoids |
Table 2: Ligands used in the study and their properties.
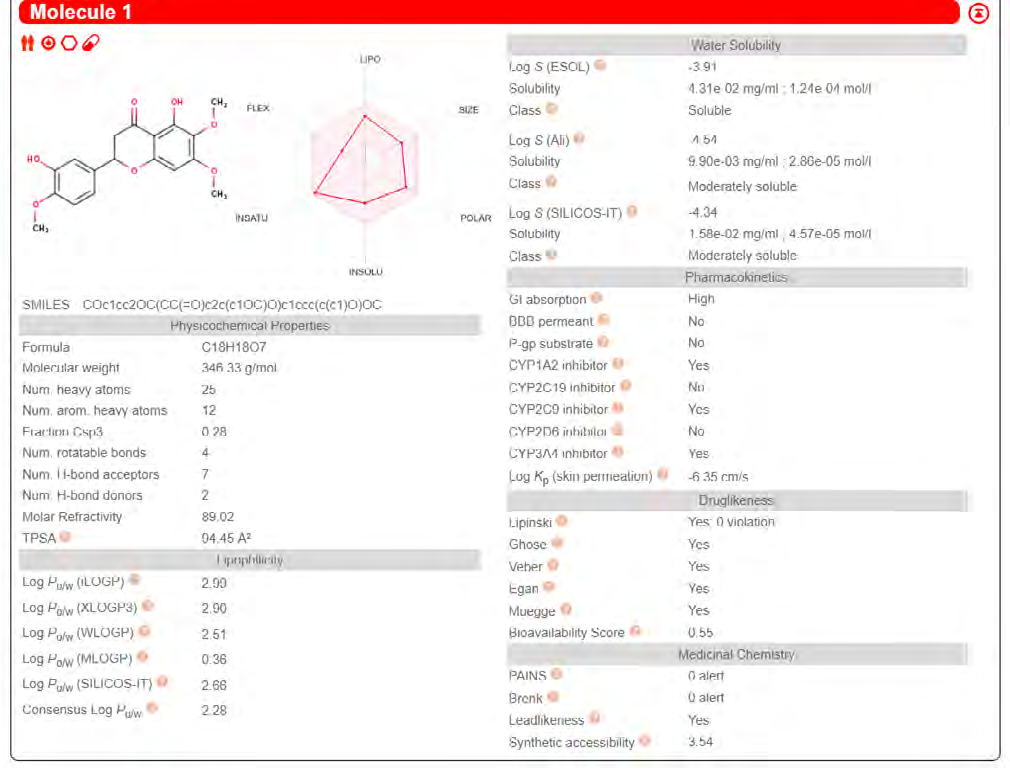
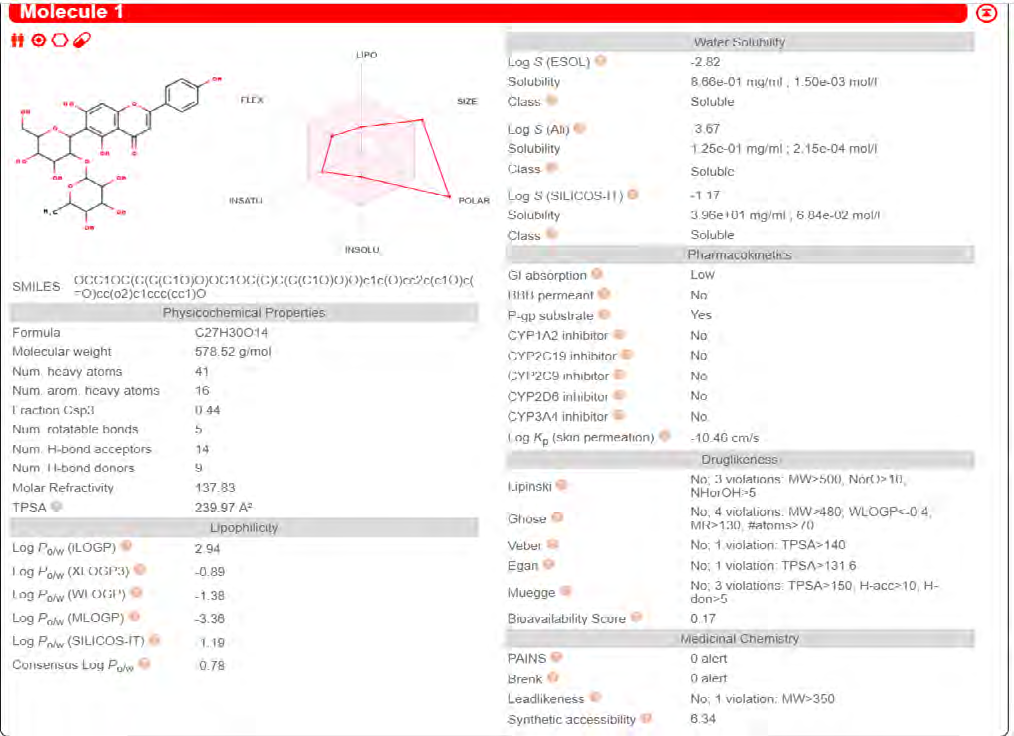
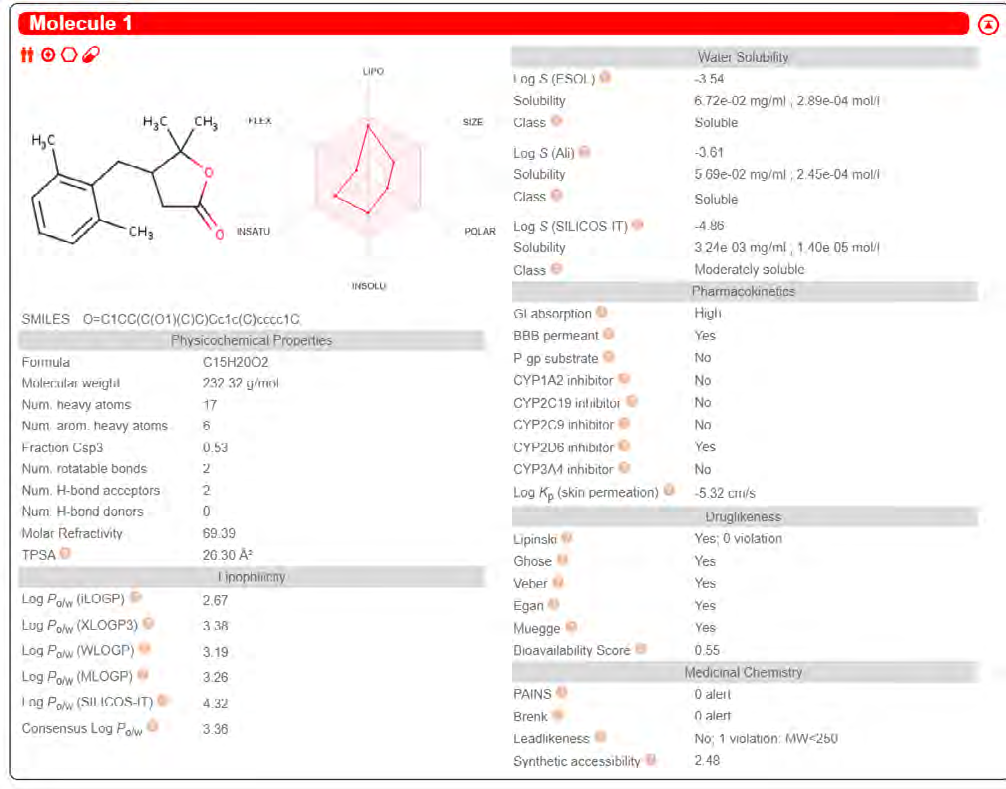
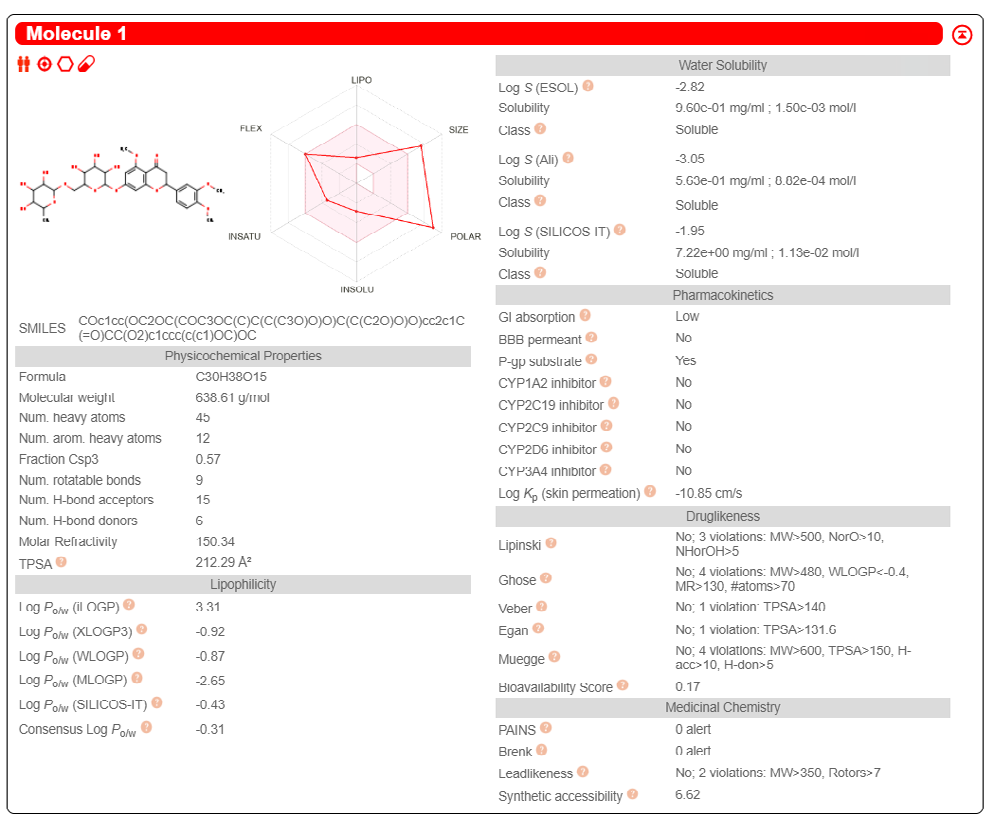
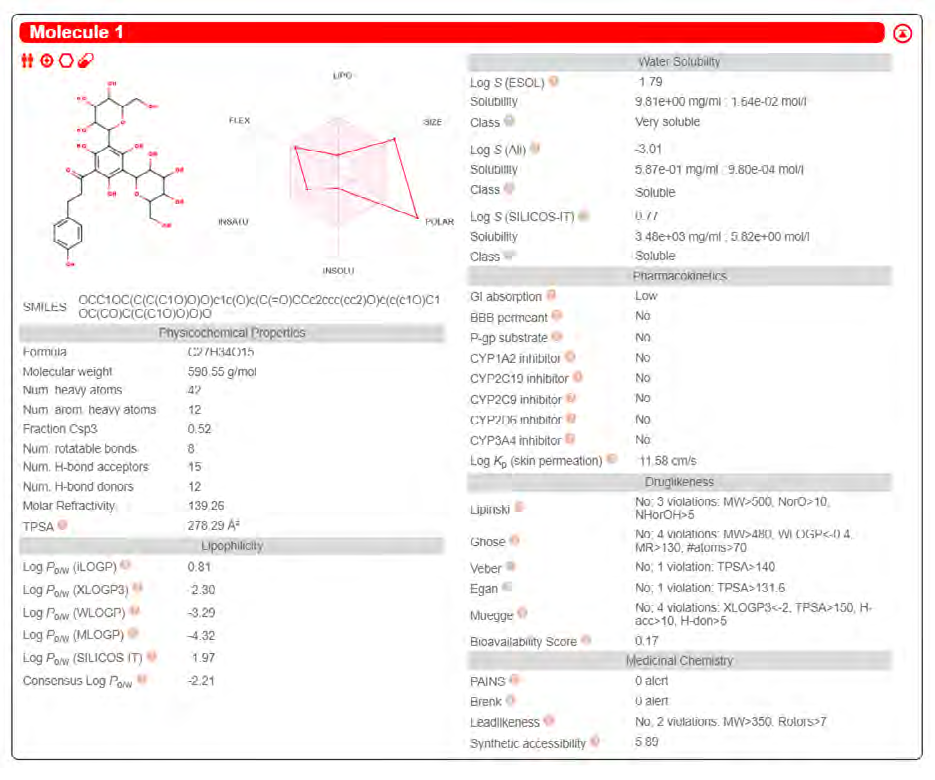
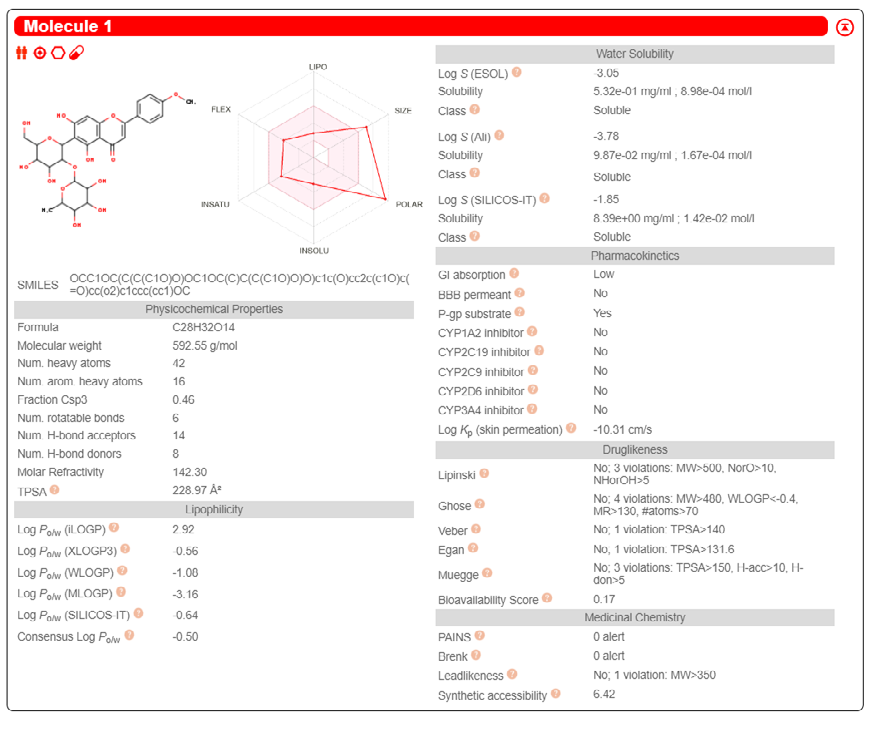
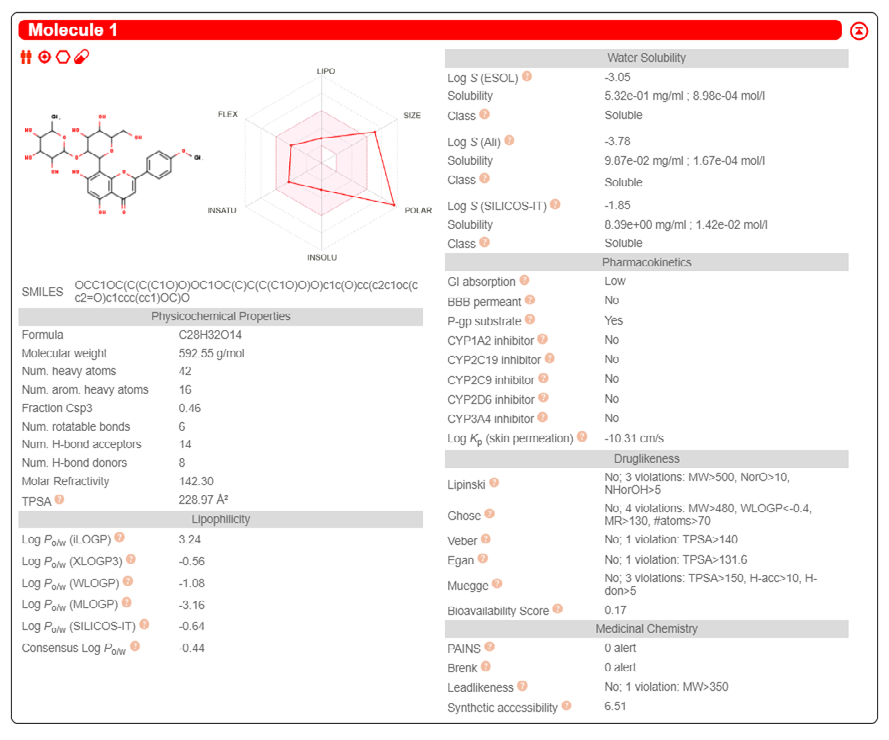
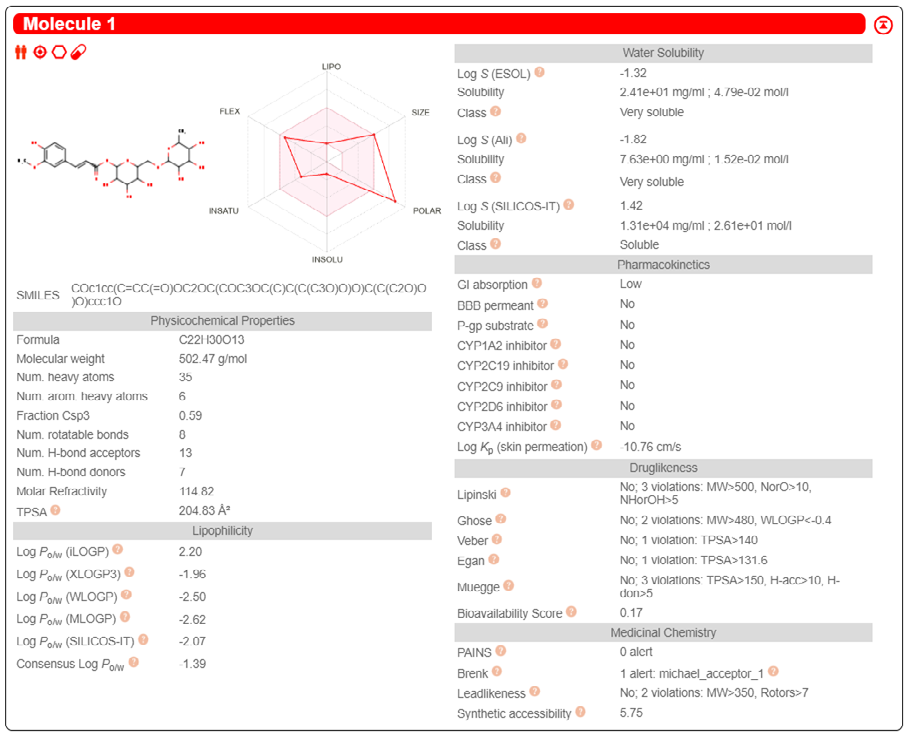
Conclusion
A research study was conducted involving the use of molecular docking and simulation techniques to explore the drug potential of various active substances in the context of SARS-CoV-2, the virus responsible for the COVID-19 pandemic. These techniques include proteases, enzymes that play a crucial role in viral replication and infection of different molecules. It is widely used in contemporary medicine and molecular biology to understand how it interacts with biological targets. The study identified eight active substances that may have the potential to act as drug candidates for the treatment of COVID-19. Using molecular docking and simulation software, the researchers analyzed the interactions between these active ingredients and SARS- CoV-2’s proteases. These in silico studies allow for virtual evaluation of the binding affinities, structural compatibility, and potential efficacy of compounds as antiviral agents. After in silico analysis, the next steps may enable in vivo and in vitro testing to confirm the findings and evaluate the actual efficacy and safety of the compounds in living organisms and biological systems. These studies will be necessary to determine whether the identified active substances can effectively inhibit viral proteases and potentially serve as therapeutic agents for COVID-19. These study results may provide a reference for other scientists and medical professionals interested in studying these compounds further. It will also contribute to ongoing efforts to find effective treatments for COVID-19 and other viral diseases. However, it is important to note that in silico research is only the first steps in drug discovery and development. Subsequent in vivo and in vitro testing are required to confirm the potential of these compounds as real drug candidates, followed by rigorous clinical trials to determine their safety and efficacy in human patients.
References
-
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. Journal of advanced research 24: 91-98.
-
World Health Organization (2020) Laboratory testing for coronavirus disease ( COVID-19) in suspected human cases: interim guidance, 19 March 2020.
-
Ouassou H, Kharchoufa L, Bouhrim M, Daoudi NE, Imtara H, et al. (2020) The Pathogenesis of Coronavirus Disease 2019 (COVID-19): Evaluation and Prevention. Journal of immunology research 2020: 1357983.
-
Gorbalenya AE, Baker SC, Baric R, Groot RJD, Drosten C, et al. (2020) Severe acute respiratory syndrome-related coronavirus: The species and its viruses-a statement of the Coronavirus Study Group.
-
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, et al. (2020) Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. BioRxiv.
-
Wu F, Zhao S, Yu B, Chen YM, Wang W, et al. (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798): 265-269.
-
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798): 270-273.
-
Lam TTY, Shum MHH, Zhu H-C, Tong Y-G, Ni XB, et al. (2020) Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. BioRxiv.
-
Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, et al. (2020) Full- genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infection, Genetics and Evolution 79: 104212.
-
Lotfi M, Hamblin MR, Rezaei N (2020) COVID-19: Transmission, prevention, and potential therapeutic opportunities. Clinica chimica acta 508: 254-266.
-
Centers for Disease Control and Prevention-Public Health Image Library. Credit: Alissa Eckert, MS, Dan Higgins, MAM (Public Domain).
-
Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr (2013) Computational methods in drug discovery. Pharmacological reviews 66(1): 334-395.
-
Ghosh R, Chakraborty A, Biswas A, Chowdhuri S (2021) Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors - an in silico docking and molecular dynamics simulation study. J Biomol Struct Dyn 2020: 1-13.
-
Jain AS, Sushma P, Dharmashekar C, Beelagi MS, Prasad SK, et al. (2021) In silico evaluation of flavonoids as effective antiviral agents on the spike glycoprotein of SARS-CoV-2. Saudi J Biol Sci 28(1): 1040-1051.
-
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2): 455-461.
-
Sas EB, Yalcin S, Ercan F, Kurt M (2020) A multi- spectroscopic, computational and molecular modeling studies on anti-apoptotic proteins with Boc-D-Lys-OH. J Mol Struct 1199: 126981.
-
Yalcin S, Sas EB, Cankaya N, Ercan F, Kurt M (2019) The physical studies and interaction with anti-apoptotic proteins of 2-(bis (cyanomethyl) amino)- 2-oxoethyl methacrylate molecule.
-
Bekker H, Berendsen HJ, Dijkstra EJ, Achterop SA, Van Drunen R, et al. (1993) Gromacs: a parallel computer for molecular dynamics simulations. Phys Comput 92: 252- 256.
-
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GRO- MOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25(13): 1656-1676.
-
Bjelkmar P, Larsson P, Cuendet MA, Hess B, Lindahl E (2010) Implementation of the charmm force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theory Comput 6: 459-466.
-
Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, et al. (2010) Improved side-chain torsion potentials for the Amber ff99SB pro- tein force field. Proteins 78(8): 1950-1958.
-
James AM, Teemu M, Roland S, Szilárd P, Jeremy CS, et al. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1: 19-25.
-
Wang Z, Wang X, Li Y, Lei T, Wang E, et al. (2019) farPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics 35(10): 1777- 1779.
-
Ntie-Kang F, Lifongo LL, Mbah JA, Owono LCO, Megnassan E, et al. (2013) In silico drug metabolism and pharmacokinetic profiles of natural products from medicinal plants in the Congo basin. In Silico Pharmacol 1: 12.
-
Zoete V, Daina A, Bovigny C, Michielin O (2016) SwissSimilarity: a web tool for low to ultra high throughput ligand-based virtual screening. J Chem Inf Model 56(8): 1399-1404.
-
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1-3): 3-25.
-
Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, et al. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45(12): 2615-2623.
-
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, et al. (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483): 1260- 1263.
-
Báez-Santos YM, Barraza SJ, Wilson MW, Agius MP, Mielech AM, et al. (2014) X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. Journal of medicinal chemistry 57(6): 2393-2412.
-
Yu R, Chen L, Lan R, Shen R, Li P (2020) Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. International journal of antimicrobial agents 56(2): 106012.
-
Turner JV, Agatonovic-Kustrin S (2007) Comprehensive medicinal chemistry II.
- hMPV: Is It Another Covid-19 Like Situation?
- Streptomyces: Sources of Novel Discoveries in Antibiotic Research to Combat Antimicrobial Resistance
- A Review of Mosquitoes (Diptera: Culicidae) and Their Biodiversity, Medical and Veterinary Importance
- Past and Current Immunotherapy in Cancer
- Hematological Cancer and Viral Infection
- The Growing Threat of Antimicrobial Resistance in India: Challenges and Solutions