In silico Approach to Retrieval of Multi-Drug Resistance Genes of Mycobacterium Tuberculosis Using Complete Genome Sequencing Technology
This study retrieved antibiotics resistance genes (ARG) information on seventy NCBI retrieved complete genomes of M. tuberculosis and their corresponding accession numbers and locations were noted, and sorted and resolved as Perfect and Strict Resistance genes. The highest number of Mycobacterium tuberculosis complete genome sequences were retrieved from the America, Africa, Antarctica, Asia, Europe and with 38%, 29%, 23% 6% and 4% respectively. The prevalence of resistant genes were categorized appropriately into their respective RGIs: These genes are: mdsB, mdsA, AAC(6’)-Iaa, sdiA, TEM-1, adeK, adeI, ADC-6, abeS, OXA-67, tetW , FTU-1, BPU-1, ANT(4’)-Ib, smeD, tet(K), tetM, mecI, ANT(4’)-Ib, KpnE, SHV-1, Oqxa had 100% prevalence each in all the 70 complete genome sequences retrieved, while golS and mecR1 had 50% prevalence. Others were: efpA, mtrA, mfpA, AAC (2’)-Ic, LmrS, ErmA with 33.3% prevalence. Both norA and FosB had 25% while arlS, mepR, mgrA had the prevalence of 16.7%, 14.28%, and 9.1% respectively. The prevalence of genes categorized under the strict category were: APH(6)-Id, APH(3’’)-Ib, sul2, tet(B), ANT(3’’)-Iic, adeJ, adeL, AmvA, adeN, adeR, AbaF, KpnH, gyrB, bacA, vanI, farB, mecR1, tet(45) ,cat86, rpsL, pncA, soxR, patB, oqxA, cmH-1, FosA2, ramA, parC, KpnG, OmpK37, FosA6, smeR, iri with 100% each. Similarly, mdfA, AbaQ, gyrA, thyA, kasA, AAC(6’)-Iy, emrB, Bla2, Bla1, mphL, arlR, blaZ, RbpA, mepR, marR 50% each. Other genes folC, acrB, acrA, kdpE, KpnF, MdtK, folP, adeF. EF-Tu, RbpA, rpoB, katG, sdiA, GlpT, uhpT 33.33% each. Five genes were 25% while two were in 20% each.
Introduction
Genomics and bioinformatics are increasingly contributing to our understanding of infectious diseases caused by bacterial pathogens such as Mycobacterium tuberculosis. Bioinformatics and WGS technology process and for optimal clinical Genomics and bioinformatics have contributed immensely to our understanding of infectious diseases: from disease pathogenesis, mechanisms and the spread of antimicrobial resistance, to host immune responses. The direct benefit of whole-genome sequencing (WGS) is the ability to provide organism identification, strain relatedness and drug resistance profile for resistance due to mutation. Comprehensive Antibiotic Resistance Database is a biological database that collects and organizes reference information on antimicrobial resistance genes, proteins and phenotypes [1]. The database covers all types of drug classes and resistance mechanisms and structures its data based on an ontology. The CARD database was one of the first resources that covered antimicrobial resistance genes [2]. Owing to advances in technology and reductions in cost, whole-genome sequencing (WGS) has been transformed from a centralized service used by a select few to integrate single genome into a relatively decentralized laboratory technique used by many to detect and track infectious pathogens [3]. Drug-resistant tuberculosis poses a significant challenge to tuberculosis control programmes in high burden settings. Undiagnosed drug resistance leads to further transmission, poor patient outcomes and potential for amplification of drug resistance which are seeming impediment to the World Health Organization’s (WHO) strategy to end tuberculosis by 2035. The drug-resistant tuberculosis outbreaks in Tugela Ferry and other regions of South Africa highlight the need for early and accurate diagnosis of drug resistance [4]. Whole Genome Sequencing (WGS) has become the reference microbial typing method in outbreak studies and is increasingly being applied to national surveillance of infectious diseases in European Union/European Economic Area (EU/EEA) countries and beyond. In 2015, European Centre for Disease Prevention and Control (ECDC) developed an expert opinion on WGS in consultation with multidisciplinary experts. European Food Safety Authority (EFSA) and research and development projects to define the priorities and identify high impact diseases or drug resistance issues, where genome sequence information can make a difference for public health intervention [5, 6].
According to the recent WHO report, there were an estimated 10.0 million cases of TB and 1.3 million deaths during the year 2017. India alone accounted for 24% of global MDR-TB incidence and 27% of global TB incidence among HIV-negative individuals (Global Tuberculosis Who Report [GWTR] x, 2018). There is an urgent need for improved diagnosis of TB, such as identification of markers to monitor transmission and effective treatment to deal with this deadly disease. Whole genome sequencing (WGS) studies from across the globe have revealed genetic diversity of Mycobacterium tuberculosis and have provided significant insights into its evolution and transmission [7]. Several studies have shown association of the genetic variations with pathogenesis and drug resistance [8]. Global frontline molecular diagnostics such as line probe assays and Xpert MTB/RIF used for diagnosis of drug resistant TB, have been developed based on these genetic markers [9, 10, 11].
Current recommendations for the treatment of drug- susceptible TB include a 6-month course of a multi-drug regimen of rifampicin, isoniazid, pyrazinamide, and ethambutol. Historically, treatment of MDR- or XDR-TB involved the long-term use of second-line drugs, including injectable agents (WHO, 2016). More recently, the MDR-TB treatment landscape has changed with the introduction of multiple novel second-line drugs that can be administered orally. In 2012, bedaquiline, adiarylquinolone, became the first TB drug from a novel drug class to receive US Food and Drug Administration (FDA) approval in over 40 years [12]. Another oral agent, delamanid, a nitro-dihydro- imidazooxazole derivative, has also shown promise for TB treatment [13]. WHO published updated treatment guidelines for MDR/RR-TB [6], recommending fully oral MDR regimens for many patient groups. Recommended treatment strategies include both shorter, standardized MDR regimens (for 9 to 12 months) and longer, individualized treatment regimens (for 18 to 20 months). The updated guidelines group anti-tubercular drugs on the basis of how they should be combined to create individualized, longer MDR-TB regimen [6, 12, 13].
Prolonged treatment courses, leading to greater drug exposure, toxicity, and non-compliance; unacceptable side- effect profiles; logistics of drug access; and re-infection [14] are great treatment challenge to TB. The dawning of the new age of genome sequencing began to revolutionize our approach to human diseases, including TB. In 1998, [15] reported the complete genome sequence of the M. tuberculosis reference strain H37Rv, which was approximately 4.41 million base pairs in length and encoded approximately 4000 genes. The first sequencing of a clinical reference strain, CDC1551, quickly followed [16]. An accompanying editorial optimistically stated: “After several decades in the slow lane of classical microbiology, M. tuberculosis is once again at the cutting edge of science”. However, even at the time of these breakthroughs, there was recognition that translating these genomic data into clinical benefit would prove challenging [17]. Despite these challenges, it is clear, more than 20 years later, that M. tuberculosis genomic data have been remarkably useful in improving our understanding of how drug-resistant TB evolves and spreads and in helping to inform diagnostics and therapies [18, 19].
Materials and Methods
Retrieval of Complete Genome Sequence of Mycobacterium Tuberculosis
A total of 70 different complete genome sequences (FASTA format) of Mycobacterium tuberculosis were retrieved from NCBI nucleotide database.
Detection of Antibiotic Resistant Genes in Mycobacterium Tuberculosis
The complete genome sequences of Mycobacterium tuberculosis were analyzed to detect the presence or absence of antibiotic resistant genes and mutants Analysis were carried out using the Comprehensive Antibiotic Resistance Database (CARD).The Resistant Gene Identifier (RGI) was employed for detection of the resistant genes and mutants present. The AMR genes were categorized as perfect and strict.
Results
Distribution of Complete Genome Sequences Of Mycobacterium Tuberculosis Per Location
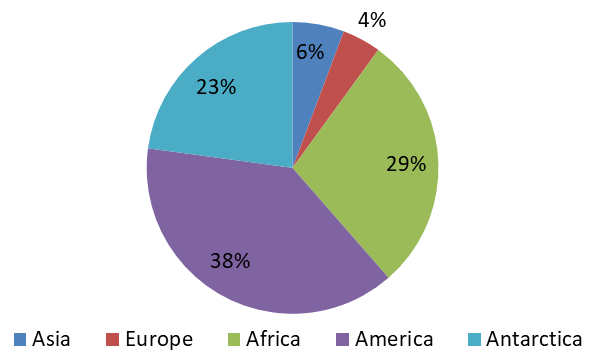
The locations from which the whole genome of Mycobacterium tuberculosis sequences retrieved for this study was shown in Figure 1. The highest number of Mycobacterium tuberculosis complete genome sequences were retrieved from the America followed by Africa and Antarctica with 38%, 29%, 23% respectively. The continents with the least number of sequences are Asia and Europe with 6% and 4% respectively.
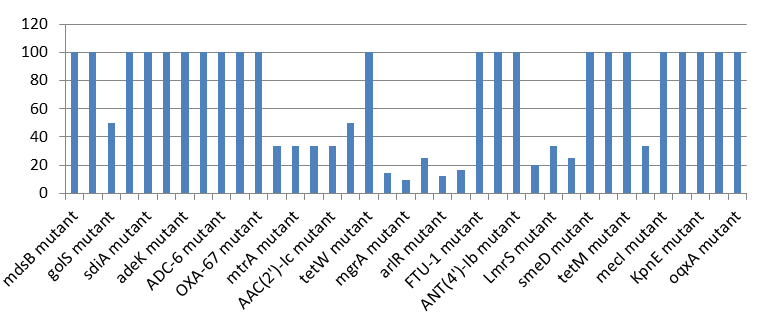
Prevalence of ‘Perfect’ Resistant Genes Present in the Complete Genome of Mycobacterium Tuberculosis Sequences.
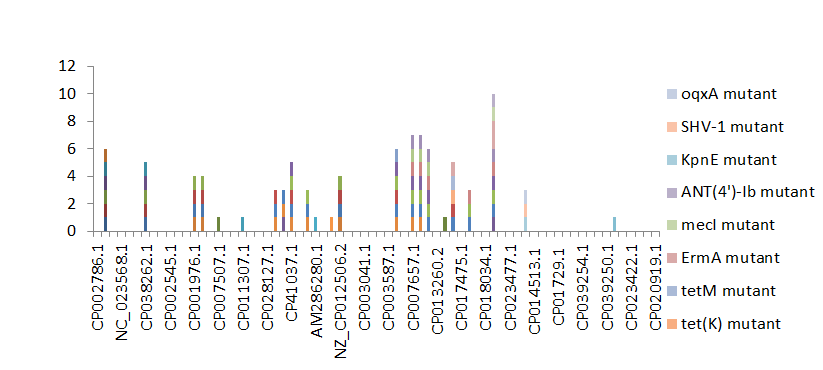
Figure 2 shows the prevalence of resistant of genes categorized under ‘perfect’ as obtained after analysis using the CARD (Comprehensive Antibiotic Resistance Database) resistant genes identifier. mdsB, mdsA, AAC(6’)-Iaa, sdiA, TEM-1, adeK, adeI, ADC-6, abeS, OXA-67, tetW , FTU-1, BPU- 1, ANT(4’)-Ib, smeD, tet(K), tetM, mecI, ANT(4’)-Ib, KpnE, SHV-1, oqxA had100% prevalence each (present in all the 70 complete genome sequences retrieved), golS and mecR1 had 50% prevalence, efpA, mtrA, mfpA, AAC(2’)-Ic, LmrS, ErmA had 33.3% prevalence each, norA and FosB had 25% prevalence each. arlS, mepR, mgrA had the prevalence rates of 16.7%, 14.3 %, and 9.1% respectively. The presence of genes under the ‘perfect’ category from CARD-RGI analysis in each of the whole genome sequences represented by their accession numbers is shown in Figure 3.
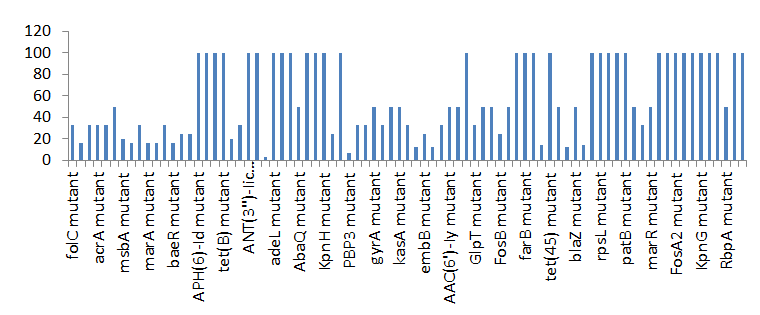
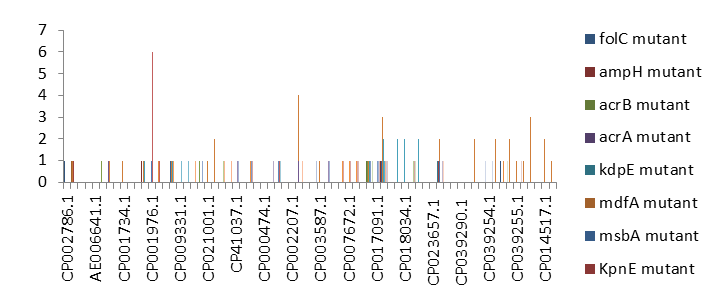
Percentage Distribution of ‘Strict’ Resistant Genes Present in the Complete Genome of Mycobacterium Tuberculosis Sequences
Figure 4 shows the prevalence of genes categorized under the strict category from CARD database with APH(6)- Id, APH(3’’)-Ib, sul2, tet(B), ANT(3’’)-Iic, adeJ, adeL, AmvA, adeN, adeR, AbaF, KpnH, gyrB, bacA, vanI, farB, mecR1, tet(45) ,cat86, rpsL, pncA, soxR, patB, oqxA, cmH-1, FosA2, ramA, parC, KpnG, OmpK37, FosA6, smeR, iri(100%) each.
mdfA, AbaQ, gyrA, thyA, kasA, AAC(6’)-Iy, emrB, Bla2, Bla1, mphL, arlR, blaZ, RbpA, mepR, marR (50%)each. folC, acrB, acrA, kdpE, KpnF, MdtK, folP, adeF. EF-Tu, RbpA, rpoB, katG, sdiA, GlpT, uhpT(33.33%) each. PmrF, ampC1, CRP, embB, FosB (25%) each. msbA and emrR had(20%)each. ampH, KpnE, marA, H-NS, baeR(16%) each. mecA and norA(14%) each. murA, rRNA and LmrS(12%)each. The presence of genes under the ‘strict’ category from CARD-RGI analysis in each of the whole genome sequences represented by their accession number is shown in Figure 5.
Discussion
Comprehensive Antibiotic Resistance Database resistant genes identifier (CARD-RGI) have been successfully used in this study to understand antibiotic resistance genes in about 70 complete genome sequences and accessions of Mycobacterium tuberculosis retrieved from the NCBI. In this study, the NCBI database was used to retrieve 70 complete genome sequences of Mycobacterium tuberculosis and the CARD RGI was used to understand what antibiotic resistance genes are present in theses genome. The lowest number of complete genome sequences retrieved from Asia and Europe, (6% and 4% respectively) due to the limited number of the complete genome sequences of Mycobacterium tuberculosis deposited in the NCBI GenBank from these locations. Drug resistance in M. tuberculosis is the result of chromosomal mutations in existing genes that are passed along through vertical descent, which is, passed from mother to daughter cells. Unlike many other bacterial pathogens, M. tuberculosis rarely recombines via lateral exchange of DNA [1] and also lacks plasmids. Many of the resistance determinants were discovered before the sequencing of the M. tuberculosis genome was completed.
The resistance genes were sorted into two; which were perfect and strict. In all, there were thirty-seven (37) perfects and Eighty-one(81) stricts APH(6)-Id mutant, APH(3’’)-Ib mutant, sul2 mutant, tet(B) mutant, ANT(3’’)-Iic mutant, adeJ mutant, adeL mutant, AmvA mutant, adeN mutant, adeR mutant, AbaF mutant, KpnH mutant, gyrB mutant, bacA mutant, vanI mutant, farB mutant, mecR1 mutant, tet(45) mutant ,cat86 mutant, rpsL mutant, pncA mutant, soxRv mutant, patB mutant, oqxA mutant, cmH-1 mutant, FosA2 mutant, ramA mutant, parC mutant, KpnG mutant, OmpK37 mutant, FosA6 mutant, smeR mutant, iri mutant, mdfA mutant, AbaQ mutant, gyrA mutant, thyA mutant, kasA mutant, AAC(6’)-Iy mutant, emrB mutant, Bla2 mutant, Bla1 mutant, mphL mutant, arlR mutant, blaZ mutant, RbpA mutant, mepR mutant, marR mutant, folC mutant, acrB mutant, acrA mutant, kdpE mutant, KpnF mutant, MdtK mutant, folP mutant, adeF mutant, EF-Tu mutant, RbpA mutant, rpoB mutant, katG mutant, sdiA mutant, GlpT mutant, uhpT mutant, PmrF mutant, ampC1 mutant, CRP mutant embB mutant, FosB mutant, msbA mutant, emrR mutant, ampH mutant, KpnE mutant, marA mutant, H-NS mutant, baeR mutant, mecA mutant, norA mutant, murA mutant, rRNA mutant, LmrS mutant, AAC(6’)-Iaa mutant, mtrA mutant, AAC(2’)-Ic mutant, SHV-1 mutant, efpA mutant, tetM mutant, tet(K) mutant, FTU-1 mutant, BPU-1 mutant, ANT(4’)-Ib mutant, smeD mutant, mecI mutant, ADC-6 mutant, abeS mutant OXA-67 mutant, mdsB mutant, mdsA mutant, TEM-1 mutant, adeK mutant, adeI mutant, tetW mutant , ANT(4’)-Ib mutant, golS mutant, mecR1 mutant, ErmA mutant, mfpA mutant, have been successfully identified. The drug-resistant tuberculosis outbreaks in Tugela Ferry and other regions of South Africa highlighted the need for early and accurate diagnosis of drug resistance [7]. Several studies have shown association of the genetic variations with pathogenesis and drug resistance [8]. Global frontline molecular diagnostics such as line probe assays and Xpert MTB/RIF used for diagnosis of drug resistant TB, have been developed based on these genetic markers [9]. However, these tests rely on a limited number of mutations. There have been several instances where phenotypic resistance could not be explained by known mutations associated with drug resistance [10]. A recent study comparing the efficacy of Xpert MTB/RIF with line probe assay for detection of rifampicin mono-resistant M. tuberculosis reported the utility of country specific probes, to increase the sensitivity of Xpert MTB/RIF in India [11]. Since there is considerable genetic heterogeneity among M. tuberculosis isolates from different geographic regions, large-scale sequencing efforts are required to map genetic variations and identify the genotypes associated with drug resistance.
Conclusion
Whole Genome Sequence investigations have revealed that there is a specific order in which drug-resistance mutations are acquired: isoniazid resistance is almost always acquired before rifampicin resistance, which has significant implications for the design of diagnostic tests. Within individual patients, WGS studies have highlighted that mixed infections are common, and often represent important intermediates in the evolution of drug resistance. Whole Genome Sequence is expected to continue to advance our understanding of M. tuberculosis drug resistance. Furthermore, its practical use in clinical settings holds great potential to improve public health through real- time molecular epidemiology tracking, to identify global hotspots of drug-resistance emergence, and to facilitate the development of improved approaches for the diagnosis and treatment of drug-resistant Mycobacterium tuberculosis.
References
-
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, et al. (2017) CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Research 45(1): D566-573.
-
Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE (2012) The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: Perhaps more? Biochimie 94(1): 110-116.
-
Long SW, Beres SB, Olsen RJ, Musser JM (2014) Absence of patient-to-patient intra hospital transmission of Staphylococcus aureus as determined by whole-genome sequencing. mBio 5(5).
-
Huson DH, Scornavacca C (2012) Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61(6): 1061-1067.
-
World Health Organization (2018) Global Tuberculosis Report, Geneva: WHO; 2018.
-
World Health Organization (2016) WHO treatment guidelines for drug-resistant tuberculosis update. Geneva: WHO; 2016.
-
Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, et al. (2014) Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet 46(3): 279-286.
-
Laurenzo D, Mousa SA (2011) Mechanisms of drug resistance in Mycobacterium tuberculosis and current status of rapid molecular diagnostic testing. Act a Trop 119(1): 5-10.
-
Gagneux S, Small PM (2007) Global phytogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 7(5): 328-337.
-
Rigouts L, Gumusboga M, De Rijk WB, Nduwamahoro E, Uwizeye C, et al. (2013) Rifampin resistance missed in automated liquid culture system for Mycobacterium tuberculosis isolates with specific rpoB mutations. J Clin Microbiol 51(8): 2641-2645.
-
Rufai SB, Kumar P, Singh A, Prajapati S, Balooni V, et al. (2014) Comparison of Xpert MTB/RIF with line probe assay for detection of rifampin-mono resistant Mycobacterium tuberculosis. J Clin Microbiol 52(6): 1846-1852.
-
Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, et al. (2012) Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug- resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrobl Agents Chemother 56(6): 3271-3276.
-
Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, et al. (2006) OPC-67683, a nitro-dihydro- imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3(11).
-
Bloom BR, Atun R, Cohen T, Dye C, Fraser H, et al. (2017) Tuberculosis In: Holmes KK, Editors. Major infectious diseases. Washington DC: The International Bank for Reconstruction and Development/The World Bank.
-
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537-544.
-
Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, et al. (2002) Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol 184(19): 5479-5490.
-
Young DB (1998) Blueprint for the white plague. Nature 393(6685): 515-516.
-
Cabibbe AM, Walker TM, Niemann S, Cirillo DM (2018) Whole genome sequencing of Mycobacterium tuberculosis. European Respiratory Journal 52: 1801163.
-
Casali N, Broda A, Harris SR, Parkhill J, Brown T, et al. (2016) Whole genome sequence analysis of a large isoniazid-resistant tuberculosis outbreak in London: a retrospective observational study. PLoS Medicine 13(10).
- Carbon Code for Analysis of Protein Stability in Protein Mutation
- Number of Contiguous Amino Acids in Nanon of 16A Diameter
- Identification of Hub Genes and Pathways in Cervical Cancer by Statistical and Bioinformatics Analysis
- Effect of Dietary Inclusion Levels of Moringa Olerifera Oil on the Growth Performance and Nutrient Retention of Broiler Starter Chicks
- Proteomics Loans in Kinetoplastids during the Last Decade
- “Identification of SARS-CoV-2 in Human Genome based on Protein Dynamics Conversion and Target Genes Marking via Bioinformatics Approaches”