PAPA like Syndrome Associated with Takayasu Arteritis Responding to Corticosteroids: A Case Report
Pyoderma gangrenosum, pyogenic sterile arthritis and acne (PAPA syndrome) is a hereditary, autosomal dominant, autoinflammatory disease caused by missense mutation in the proline/serine/threonine phosphatase-interacting protein 1 (PSTPIP1) gene and predominantly affecting the joints and skin. Many reports now suggest that the clinical phenotype of PAPA syndrome may vary among people carrying the mutation and can lack the classic triad of pyogenic sterile arthritis, Pyoderma gangrenosum and cystic acne. No definite treatment strategy has been established for this PAPA like syndrome so far. We describe the treatment response of corticosteroids in a 23-year-old male with PAPA like syndrome and Takayasu arteritis, an association which has never been reported before.
Manuscript
PAPA (Pyoderma gangrenosum, pyogenic sterile arthritis and acne) syndrome is a hereditary, autosomal dominant, auto-inflammatory disease caused by missense mutation in the proline/serine/threonine phosphatase-interacting protein 1(PSTPIP1) gene. This condition predominantly affects the joints and skin. The clinical phenotype of PAPA syndrome may vary to the point of lacking its classic triad and is henceforth referred to as PAPA-like syndrome [1]. We describe 23-year-old man with PAPA-like syndrome and Takayasu arteritis, an association which has never been reported before and its favourable response to systemic corticosteroids.
A 23-year-old male presented with red raised pus filled lesions on his face, neck, upper chest, shoulders and back, on and off for three years and painful ulcer over his neck of one month’s duration. There were spells of fever (upto 101°F) accompanied by pain in lower back and both knees. On examination, polymorphic lesions of acne; comedones, erythematous papules and pustules, nodules and post acne scarring were noted. A single painful ulcer mounted with adherent hemorrhagic crusting was noted covering almost the entire neck anteriorly and posteriorly (Figure 1). Removal of crust revealed purulent, foul smelling, yellowish coloured discharge along with necrotic slough on the floor (Figure 2). Retroauricular areas, axillae and inguinal folds were clear. Mucosa, hair and nail examination was normal. There was no erythema or swelling on joint examination. Interestingly, the blood pressure in his right arm was 120/78 mm Hg with good volume but in the left arm was 90/70 mm Hg with feeble pulse. Review of systems was normal. Investigations of the patient are summarized in Table 1. Based on the clinical and histopathological findings of Pyoderma gangrenosum and acne and the absence of pyogenic arthritis, diagnosis of PAPA-like syndrome was rendered. Association with Takayasu arteritis was suggested by angiographic findings. Patient was treated as an inpatient with local wound care, non-steroidal anti-inflammatory drugs (NSAIDs), systemic steroids- initially injectable dexamethasone 4 mg once daily and prophylactic antibiotic- injectable amoxicillin- clavulanate 1.2 gm twice daily for 7 days followed by tab prednisolone 50 mg and cap doxycycline 100 mg twice daily along with iron, calcium and vitamin D3 supplementation. There was complete clearing of skin lesions and improvement in general well-being of patient within four weeks (Figure 3). Patient is under regular follow up.
| Hemoglobin | 9.4 mg/dl | Tzanck smear | Lypmhoneutrophilic infiltrate only |
|---|---|---|---|
| TLC | 11,400/mm3 (75% neutrophils) | Direct Immunofluorescence | Negative |
| Platelet count | 2,80,000/ul | Pus culture | Negative |
| LFT | Normal | Blood culture | Negative |
| KFT | Normal | Ziehl Neelsen stain for AFB | Negative |
| Urine routine microscopy | Normal | Culture for Acid fast bacilli | Negative |
| CRP | Positive | KOH mount | Negative |
| Viral markers | Negative | Fungal culture | Negative |
| ANA, Rheumatoid factor | Negative | Histopathology (from ulcer) | Focal areas of ulceration in epidermis and diffuse dense neutrophilic infiltrate in the dermis |
| Mantoux test | Negative | Histopathology (from nodulocystic lesion) | Follicular occlusion and inflammation with evidence of suppurative granulomas in the perifollicular area |
| Chest X ray | Normal | ECG | Normal |
| X ray knees and lumbosacral spine | Mild osteopenia | ECHO | Normal |
| PSTPIP1 gene analysis | CT angiography (left limb) | Large vessel vasculitis, likely Takayasu arteritis | |
| Not done due to non-availability |
Table 1: Table summarizing the investigations.
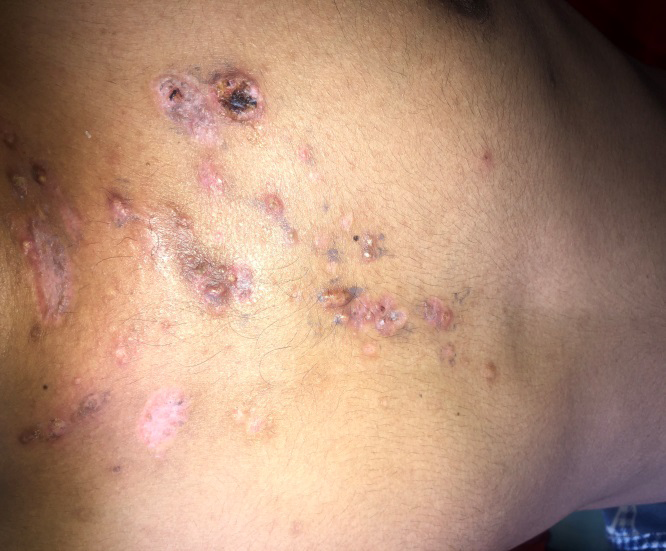
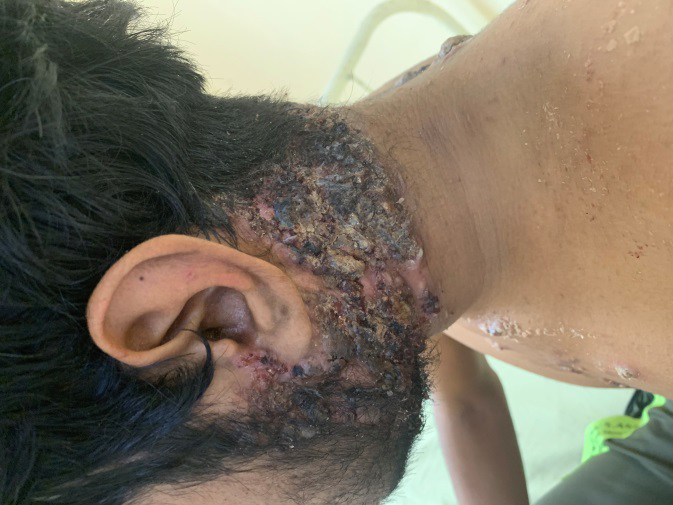
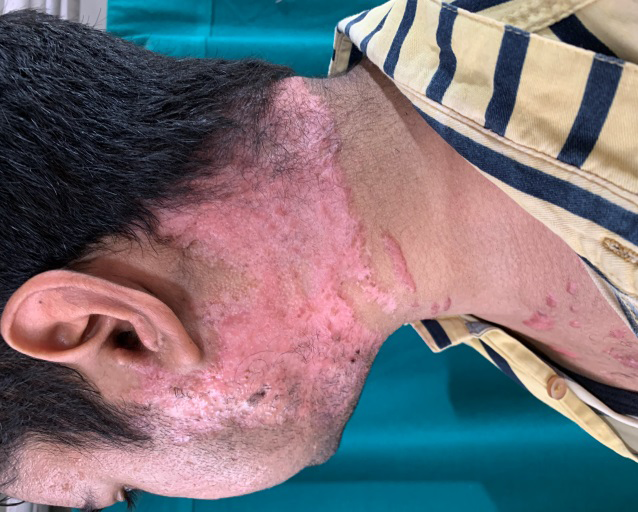
PAPA syndrome is an auto-inflammatory disease whose clinical phenotype may vary amongst the people carrying the PSTPIP1 gene mutation and can lack either of the three classic features or have a late onset [1]. Nesterovitch et al. reported isolated Pyoderma gangrenosum associated with mutation in PSTPIP1 gene [2]. Another case series described three females of the same family, 6 year old girl having arthritis and pustulosis; her 33 year old mother having arthritis, acne and abscesses; and 48 year old cousin having arthritis and acne. Pyoderma gangrenosum was absent in all three [3]. Our patient presented in adulthood and had Pyoderma gangrenosum along with acne but lacked pyogenic arthritis. Additionally, our patient had Takayasu’s arteritis. Association of PAPA-like syndrome with ulcerative colitis was reported in a 33-year-old male by which responded well to Anakinra [4]. To our knowledge, this is the first reported case of PAPA-like syndrome with Takayasu arteritis. No standard treatment strategy for PAPA like syndrome has been established; nonetheless, rapid remission was achieved in a
22-year-old Iranian male having Pyoderma gangrenosum and acne fulminans with canakinumab, anti–interleukin 1 beta monoclonal antibody. 1 Takayasu arteritis associated with Pyoderma gangrenosum has been widely and successfully treated with systemic corticosteroids [5].
To conclude, PAPA and PAPA like syndromes are hereditary, autoinflammatory syndromes resulting from variable phenotypic expression of PSTPIP1 gene. This is the first reported case of PAPA like syndrome associated with Takayasu arteritis which responded well to corticosteroids.
References
-
Geusau A, Mothes-Luksch N, Nahavandi H, Pickl WF, Wise CA, et al. (2013) Identification of a homozygous PSTPIP1 mutation in a patient with a PAPA-like syndrome responding to canakinumab treatment. JAMA Dermatol 149(2): 209-215.
-
Nesterovitch AB, Hoffman MD, Simon M, Petukhov PA, Tharp MD, et al. (2011) Mutations in the PSTPIP1 gene and aberrant splicing variants in patients with _Pyoderma_ _gangrenosum_. Clin Exp Dermatol 36(8): 889-895.
-
Schellevis MA, Stoffels M, Hoppenreijs EP, Bodar E, Simon A, et al. (2011) Variable expression and treatment of PAPA syndrome. Ann Rheum Dis 70(6): 1168-1170.
-
Zeeli T, Padalon-Brauch G, Ellenbogen E, Gat A, Sarig O, et al (2015) _Pyoderma gangrenosum_, acne and ulcerative colitis in a patient with a novel mutation in the PSTPIP1 gene. Clin Exp Dermatol 40(4): 367-372.
-
Ujiie H, Sawamura D, Yokota K, Nishie W, Shichinohe R, et al. (2004) _Pyoderma gangrenosum_ associated with Takayasu’s arteritis. Clin Exp Dermatol 29(4): 357-359.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling