Recent Approaches of Impurity Profiling in Pharmaceutical Analysis: A Concise Review
Purity profiling is the process of gathering and analyzing information to determine the biological safety of a specific impurity, hence highlighting its importance and range in pharmaceutical research. In the field of pharmaceuticals, impurity has no precise meaning. Identification, structural elucidation, and quantitative determination of impurities and degradation products in bulk medicinal materials and pharmaceutical formulations are all included in impurity profiling. Since unrecognized, possibly poisonous impurities are dangerous to health and should be found and determined by selective procedures in order to increase the safety of drug therapy, impurity profiling has become more significant in contemporary pharmaceutical analysis. Impurities are typically described using words like residual solvents, byproducts, transformation products, degradation products, interaction products, and related products. Impurity identification is carried out using different chromatographic methods in addition to passing the CGMP, QC, QA, and water activity tests, a pharmaceutical ingredient also needs to satisfy the requirements for a new impurity. It is important to separate and characterize impurities in order to collect and evaluate data that determines biological safety, which emphasizes the need for and promise of drug impurity profiling in pharmaceutical research. To separate and measure the pollutants, a range of instrumental analytical techniques have been consistently used. The detection and regulatory evaluation of organic impurities is a very challenging task because to the numerous sources of organic impurities, including microbiological contamination, API breakdown products, and traces of intermediates.
Introduction
A constant in the pharmaceutical industry is that the product should A be as pure as possible. Whether new pharmaceuticals are being developed from natural ingredients or synthetic chemically manufactured drug components, the pharmaceutical industry is continually looking to develop new drugs. Purity has therefore always been seen as a crucial component in assuring the quality of drugs there isn’t a drug on the market that isn’t dangerous or even poisonous in excessive quantities. It can be said that between the time of Paracelsus, who lived in Basel during the first half of the 16th century, and that of Ehrlich, to that of Ehrlich, to that of Ehrlich, there was a gradual shift away from the use of natural products in their entirety to either purified extracts from those products or to synthetic chemically produced substances [1].
Since it is the source of active pharmaceutical ingredients (APIs) of a certain grade, the bulk drug sector serves as the foundation of all pharmaceutical industries. The quality of new drugs entering the market has received a lot of attention during the past few decades. Producing quality products is a key difficulty for both the pharmaceutical and bulk medication sectors. To ensure the quality and purity of the product from each industry, rigorous quality control inspections must be carried out. The type of crystallization and purification procedure, as well as the raw materials used in their production, all affects the active medicinal ingredient’s purity. The idea of purity evolves over time and is inextricably linked to advancements in analytical chemistry. The pharmacopoeias set limitations that can be very strict on levels of various contaminants in addition to purity requirements. Since these procedures simultaneously separate and quantify the components, they clearly occupy a leading position in scientific research today. This makes it simpler to distinguish and characterize contaminants.
Pharmaceutical impurities are undesired substances that remain with Active Pharmaceutical Ingredients (APIs), occur during formulation, or appear as medications age [2, 3, 4, 5]. Unwanted substances that remain with the Active Pharmaceutical Ingredients (APIs), arise during formulation, or appear as medications age are known as impurities in pharmaceuticals [3, 4, 5, 6]. Drug product impurities can be incorporated into a drug product during the formulation process or by coming into touch with the packaging of the numerous impurities that can be found in drug products, in addition to the drug substance or inert materials utilized to create a drug product. “Any component of the drug product that is not a drug substance or an excipient in the drug product,” according to the definition. (ICH Q6A: Requirements) [7].
Impurities found in APIs are of ever-increasing interest. Recent regulatory requirements have made it necessary to have both a purity profile and an impurity profile. In the pharmaceutical industry, an impurity is defined as any additional organic material that results from synthesis or undesirable compounds that are still present in APIs. The impurity may appear during formulation or after both raw APIs and formed APIs have aged in pharmaceutical products. Identification of impurities in APIs such as 1-(1, 2, 3, 5, 6, 7 hexahydro-s-indacen-4-yl)-3-4[-1-hydroxy-1-methyl-ethyl]- furan-2-sulphonylurea utilizing multidisciplinary approach may serve as an effective demonstration of this description [8].
Even a little amount of these undesirable compounds can have an impact on the safety and effectiveness of medicinal goods. Regulatory agencies are now paying critical attention to impurity profiling, which involves the identification and quantification of impurities in medications. Limits to permissible amounts of impurities present in the APIs or formulations are gradually being incorporated by the various Pharmacopoeias, including the British Pharmacopoeia (BP), United States Pharmacopoeia (USP), and Indian Pharmacopoeia (IP).Guidelines for validating techniques for analyzing impurities in new drug substances, products, residual solvents, and microbiological impurities have also been published by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) [9, 10, 11, 12].
Definition of Impurity and Impurity Profiling
Impurities are compounds found in a product that are neither Active Pharmaceutical Ingredients (API) themselves nor the excipients used to create it, according to the International Conference on Harmonization (ICH) rules. While IP has defined impurity as any pharmacological substance’s component for a drug product that is not the chemical entity that is used in a pharmaceutical setting or explains the ingredient, or excipients in the case of a drug product, but not in the case of a drug product Impurity is simply any substance that coexists with the target ingredient. Initial drug, such as the precursors, intermediaries, or produced; as a result of any adverse effects. An impurity profile is a description of both known and unknown impurities. Contaminants in pharmaceutical products [13].
The following are the primary causes for the rising interest of drug producers and drug registration agencies in the impurity profiles of bulk drug compounds:
- It is crucial to understand the structures of the contaminants during the creation of a new drug or a new manufacturing method for an already-marketed drug: Synthetic organic chemists frequently have the knowledge necessary to alter the conditions of reactions in such that the impurity’s production can be prevented or its quantity can be decreased to a reasonable level.
- The impurities can be manufactured after having recommended structures for them, providing concrete proof of the structures that were previously established using spectroscopic techniques.
- When creating a selective procedure for the quantitative analysis, the material created might be utilized as an “impurity standard.” identifying the impurity and applying using this technique as a quality control measure testing of every batch.
- In the event of significant contaminants, the synthetic or solitary materials could be exposed to Thus, toxicological research significantly contributes to the security of pharmacological therapy.
- The impurity profile of a drug substance is a reliable fingerprint for drug regulators to identify the consistency and scale of the production process of the bulk drug substance [14].
Pharmacopoeial and Regulatory Guidelines and Status on Impurity Profiling
The impurity profiling of the medications was not given significant emphasis in earlier editions of several pharmacopoeias. However, in more recent editions, emphasis has been placed on impurity profiling for numerous medications and inclusion in the monograph. Limits to permissible levels of contaminants contained in API and formulations have been established by IP, BP, and USP. Guidelines for validating techniques for analyzing impurities in new drug substances, products, residual solvents, and microbial impurities have also been published by the International Conference on Harmonization (ICH) Guideline for Technical Requirements for Registration of Pharmaceuticals for Human Use [15].
According to USP, improvements in analytical chemistry are inseparable from changes in the idea of purity over time. If an item that was once thought to be pure can now be classified as having inorganic, organic, isomeric, or polymeric components that are deemed impurities. Setting pharmacopoeial standards can be difficult because completed medications might have either high or low purity levels. Where a preparation degrades with time when time is a factor, the same analytical techniques that indicate stability also indicate purity [16].
Impurities, according to BP, are separated into two subtypes called “Qualified ‘Detectable impurities’ and other ‘Impurities’. These are the eligible impurities. formerly recognized as qualified viz. Impurities by competent authorities They are present as natural metabolites, along with other “Detectable Impurities,” These were not found in any samples of the chemicals taken during the process of elaboration of the monograph or that happens in concentrations less than 0.1% but has been demonstrated being constrained by testing The pharmacopoeia’s monographs have been created to guarantee the consumers’ access to drug substances and products of a minimum acceptable standard. Numerous monographs have looked into similar substance tests to reduce contaminants and degradation byproducts. Although ensuring the identity, strength, purity, and quality of official products is one of the Pharmacopoeia’s main goals, it is not practical to include a test for every impurity, contaminant, or even adulterant that might be present in each monograph [17]. Table 1 lists the impurity acceptability standard. Impurities in drug products must be monitored for safety, efficacy, and economic as well as competitive reasons. However, even in the pharmaceutical sciences and business, monitoring contaminants and regulating these impurities might imply different things to different individuals or to the same people at different times. To ensure that everyone uses the same word when addressing queries about impurity, a combined terminology is required. The guidance created under the direction of the ICH has been approved by the US Food and Drug Administration (US- FDA). The European Union (EU), Japan, and the United States worked together to produce the ICH guideline for impurities in pharmaceuticals, which has helped to ensure that different areas have dependable standards for the data that should be reported to various regulatory bodies. The guidelines support the FDA reviewers and field investigators in their consistent interpretation and application of regulations, as well as the sponsors of New Drug Applications (NDA) or Abbreviated New Drug Applications (ANDA) with the type of information that should be submitted with their applications. The different regulatory requirements for impurities, Table 2 [13].
| for Drug Substances | for Drug Products | |
|---|---|---|
| Each identified specified impurity | 0.50% | - |
| Each unidentified impurity | 0.30% | - |
| Total impurity | 1.00% | - |
| Each identified specific degradation product | - | 1.00% |
| Each unidentified degradation product | - | 0.50% |
Table 1: Acceptance criteria for Impurities (As per Indian Pharmacopoeia).
| Depiction | |
|---|---|
| Q1A | ICH guidelines “stability testing of new drug substances and products” |
| Q3A | ICH guidelines “Impurities in New Drug Substances” |
| Q3B | ICH guidelines “Impurities in New Drug Products” |
| Q3C | ICH guidelines “Impurities: Guidelines for residual solvents” |
| US-FDA | “NDAs -Impurities in New Drug Substances” |
| USFDA | “ANDAs – Impurities in New Drug Substances” |
| Australian regulatory guideline | Australian regulatory guideline for prescription medicines, Therapeutic Governance Authority (TGA), Australia. |
Table 2: Regulatory guidelines.
Regulatory Framework for Controlling Impurities
Impurities are managed within the scope of the multidisciplinary guidance and the International Conference of Harmonization (ICH) quality recommendations (ICH Q3A, Q3B, Q3C, Q3D, and Q6A, Q6B) (ICH M3 and M7). The latter is the subject of a separate review in this issue, so it won’t be covered here.
ICH Q3A: Offers guidelines for the composition, detection, and validation of contaminants in novel APIs made through chemical synthesis. When making APIs or afterward storing them, organic contaminants can appear. Starting materials, reagents, by-products, intermediates, filter aids, and degradation products are some of the categories that they fall under. They can be either ICHQ3 identifiable or unidentified. The guidance explains API impurity categorization and identification, impurity listing in specifications (see also ICH Q6A, ICH Q6B), and pertinent analytical processes. The guidelines also provide relevant advice for identifying contaminants in batches of a novel API used in safety and clinical investigations by using the necessary safety studies. Similar steps should be taken to control other achiral impurities when attempting to manage the stereo chemical or enantiomer purity of an API.
ICHQ3B: Similar to this, ICH Q3B offers instructions on the nature, classification, and content of contaminants in new medicinal products. The scope of ICH Q3B is similar to that of ICH Q3A. Impurities in brand-new pharmaceutical medicines (or degradation products) develop during production or storage. Usually, they are degradation products of the API or reaction products of the API with a processing aid, an excipient (or an impurity inside an excipient), or the principal packaging materials that are used right away.
ICHQ3C: The ICH Q3C advises the appropriate levels of residual solvents that are permitted in APIs and medicinal formulations. The best solvent choice during an API synthesis process can influence key physicochemical properties of the API, such as crystal shape, purity, and solubility, as well as the yield of the finished product. As a result, in the course of the synthetic process, the solvent could occasionally be a critical process parameter (CPP). The recommendation outlines the allowed daily exposure (PDE) limits for various residual solvents that are regarded to be toxicologically acceptable and suggests using less toxic substitutes, such as avoiding class 1 toxic solvents like benzene. For class 3 solvents, ALARP considerations based on process capability are necessary, but they are expressly not applicable to class 2 solvents. However, in practice, regulators will anticipate that all classes of solvents will be subject to process capability evaluations.
ICHQ3D: The allowed levels of remaining elemental contaminants in medicinal products are suggested by ICH Q3D. There are various places where elemental impurities might come from. During API synthesis, catalyzed coupling reactions-often at a late stage-are the primary source of residual catalysts. By boosting yields and improving process efficiency, these strategies often lower the cost of goods (CoGs), which is a benefit to the environment. Alternately, elemental impurities may manifest as API or drug product impurities, such as leachable from primary or secondary processing machinery or from packaging materials, drug product impurities in excipients, or API impurities in a medicine’s active ingredient. The recommendations encompass the review of the toxicity data for probable elemental impurities, the establishment of PDEs (permitted daily exposures) for each elemental impurity, and the use of a risk based approach to regulate those impurities in medicinal products. The safety-based restrictions alone are regarded to be sufficient, and ALARP considerations based on process capability evaluations are not particularly required [18].
Sources of Impurity in Medicine
Formulations may contain impurities from a variety of sources. Impurities can have main three different sources: (I) impurities linked to APIs, which might result from stereochemistry, crystallization, or the functional group of APIs, and (II) impurities connected to processes. Which include the substances employed as reagents, catalysts, and compounds Impurities linked to synthesis, intermediates and byproducts, degradation products, method condition- and formulation-related impurities, and (III) impurities related to stability, such as API degradation and interactions between APIs and formulation excipients [19].
Organic Impurities
If sufficient attention is not taken in each phase of the multi- step synthesis, organic contaminants are the most prevalent impurities detected in any API. If the makers are not extremely vigilant about the impurities, even though the end products are always cleansed with solvents, it is always possible that the residual unreacted beginning ingredients will remain. There is a limit test for p-aminophenol in a paracetamol bulk, which may be used as a beginning material by one manufacturer or as an intermediate by another (Figure 2).
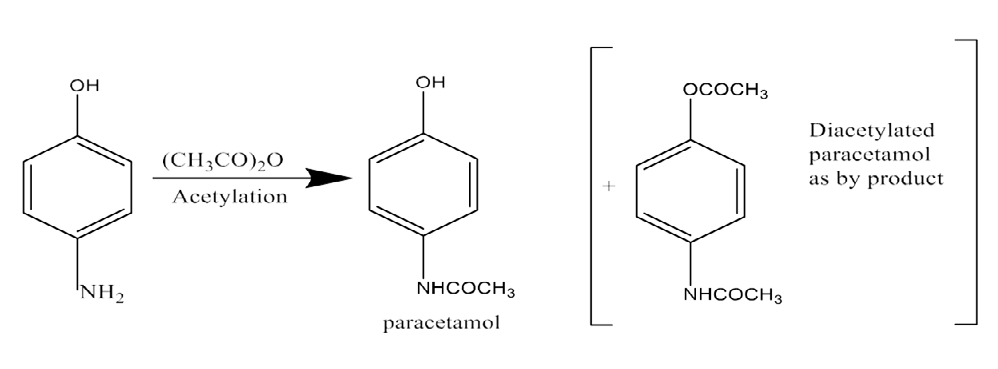
Figure 2: Paracetamol is produced from the intermediate p-Aminophenol. Oxidative degradation: The following substances are vulnerable to oxidative degradation: hydrocortisone, methotrexate, adinazolam, hydroxyl groups directly bonded to aromatic rings (e.g., phenol derivatives like catecholamines and morphine), conjugated dienes, heterocyclic aromatic rings, nitroso and nitrite derivatives, and aldehydes (e.g., flavones).
Decarboxylation: In the case of the photoreaction of rufloxacin, some dissolved carboxylic acids, such as paminosalicylic acid, lose carbon dioxide from the carboxyl group when heated in the cace of photoreaction of rufloxacin [24].
Hydrolysis: For medications of the ester type, hydrolysis is a frequent occurrence, particularly in liquid dosing forms. Barbitol, chloramphenicol, chlordiazepoxide, lincomycin, ethyl paraben [21], and cefpodoxime proxetil are a few examples [25].
Photolytic Cleavage
Pharmaceutical items are made as a solid or solution and then packaged after being exposed to light during manufacturing. When exposed to high energy UV radiation, the majority of substances breakdown as solutions (Ergometrine [26] Nifedipine [27] riboflavin, and phenothiazines are very labile to photooxidation.). It has been discovered that fluoroquinolone antibiotics are also photolytically cleavable [28]. The photocleavage reaction creates the ethylenediamine analogue of ciprofloxacin in ciprofloxacin eye drops [29].
Enantiomeric Impurities: A chiral drug’s single enantiomeric form is now regarded as a superior chemical compound with the potential to have a better pharmacological profile, a higher therapeutic index, and a more palatable adverse response profile. The pharmacokinetic characteristics of ofloxacin (R-isomeric form) and levofloxacin (S-isomeric form) are similar, indicating that there are no advantages to using a single isomer in this situation. The unwanted stereoisomers in drug control are viewed by producers of a single enantiomeric drug (eutomer) in the same way as other organic contaminants [30]. Inorganic Impurities: It’s also possible for inorganic contaminants to be produced during the manufacture of bulk medications. They include the following and are typically 11tilized 1111 and named: Catalysts, Ligands, and Reagents: These contaminants are extremely unlikely to exist, although in some procedures they might if the producers don’t take adequate precautions throughout production. Heavy Metals: When acidification or acid hydrolysis occurs, the water 11tilized in the procedures and the reactors (if stainless steel reactors are employed) are the principal sources of heavy metals. It is simple to prevent these heavy metal contaminants by 11tilized11 demineralized water and glass- lined reactors. Other Materials (Such as Charcoal, Filter Aids, etc.): Activated carbon is frequently 11tilized in bulk drug manufacturing facilities in addition to filters or filtering aids like centrifuge bags. To prevent these contaminations, it is crucial to regularly check the bulk medications for fibres and black particles.
In-Process Production Impuritie: Contaminants Connected To Crystallization: Any substance other than the one being crystallized can be an impurity. As a result, even the solvent used to create the crystals can be viewed as an impurity. Additives are impurities that are purposefully applied to generate a particular morphological impact. In a crystallisation system, the presence of impurities or additions can drastically alter crystal growth, nucleation, and agglomeration as well as the uptake of foreign ions in the crystal structure [31]. Solvents Remain after Processing: Organic volatile compounds used in manufacturing or produced during production are residual solvents. When making bulk pharmaceuticals, certain solvents that are known to be harmful should be avoided [32]. Remaining solvents are broken down into three categories based on the potential risk to human health (Table 3).
| Name of the solvent/limits | Unit/specification | |
|---|---|---|
| Class i | Benzene (2 ppm),carbon tetrachloride (4 ppm),methylene chloride (600 ppm),(methanol (3000 ppm) pyridine(200 ppm) tolune(890 ppm) | More than this should be avoided |
| Class ii | N, Ndimethyleformamide (880 ppm), acetonitride (410 ppm) | More than this should be avoided |
| Class iii | Acetic acid, ethanol, acetone (50mg) | have permitted daily exposure of 50 mg or less per day, as per the ICH guideline. |
Table 3: Classification of solvents on the basis of their limit in parts per million (ppm).
Synthetic By-Products and Intermediates Impurities: In new chemical entities (NCEs) or medicinal compounds can develop during the synthetic process from starting materials, intermediates, and/or by-products. Impurities were formed in the intermediates via the reductive amination process as a result of the impurity profile of tablets by GC-MS and MDMA (3, 4-Methylene dioxy methamphetamine) samples [33]. Impurities Generated During Storage: Numerous contaminants might develop when medication products are being stored or transported. Stability studies must be conducted in order to forecast, assess, and guarantee the safety of pharmacological products [3]. Metal Impurities: In the APIs and excipients, metal behaves as an impurity. Three categories can be used to categorise metals, as shown in Table 4 [29]. Leachables/Extractables: Regulatory, safety, and scientific considerations in evaluating extractables and leachables is important, along with strategy studies, for analytical identification, quantification, and monitoring [34].
| Category | Example | |
|---|---|---|
| Class i | metals of significant safety concern | Ir (iridinium), Pt(platinum), Rh(rhubedium), Mo(molibidnum), V(vanadium)Cr(chromium) and Ni(nickel) |
| Class ii | metals with low safety concern | Cu(copper) and Mn(manganese) |
| Class iii | metals with minimal safety concern | Fe(iron) and Zn(zinc) |
Table 4: Classification of metals on the basis of their safety concern.
Formation of Impurities on Aging
Mutual interaction amongst ingredients
The majority of vitamins is exceedingly labile and become unstable in various dose forms as people age [40]. Especially in dose formulations for liquids. When vitamins like folic acid, pantothenic acid, cyanocobalamin, and thiamine degrade, harmful contaminants are not produced; however, the potency of the active ingredients falls below pharmacopoeial specifications. The inclusion of nicotinamide in a formulation with four vitamins causes their interactions to increase (nicotinamide, pyridoxine, riboflavin, and thiamine) causes vitamin B-complex injections’ thiamine to deteriorate to an inadequate level within a year of being stored [41]. The pH of the vitamin B-complex injections that were sold had been determined to be between 2.8 and 4.0. Investigations into the specially created formulation in a straightforward distilled- water vehicle and in a conventionally formulated vehicle including disodium editate and benzyl alcohol revealed similar reciprocal interactions leading to deterioration.
Functional Group-Related Typical Degradation
Ester Hydrolysis: The following were examples. Aspirin, benzocaine, cefotaxime, cocaine echothiophate, ethyl paraben [42], cefpodoxime proxetil [43].
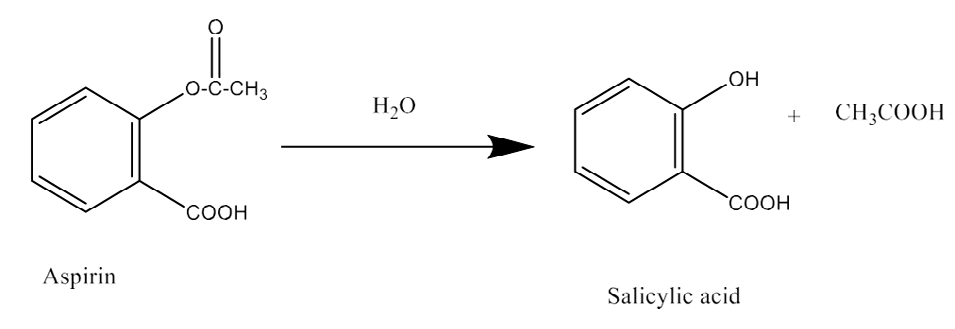
Hydrolysis: When using liquid dosage forms of ester-type medications, hydrolysis is a regular occurrence. Examples include barbitol, chloramphenicol, chlordiazepoxide, oxazepam, benzylpenicillin, and chloramphenicol [42]. Oxidative Degradation: Oxidative degradation can occur with hydrocortisone, methotrexate, adinazolam, conjugated dienes (like vitamin A and unsaturated free fatty acids), heterocyclic aromatic rings, nitroso and nitrite derivatives, aldehydes (like flavourings), and hydroxyl groups directly attached to an aromatic ring. Photolytic Cleavage: Pharmaceuticals are made as a solid or solution, packaged, and stored in pharmacies all while being exposed to light. or institutions for future use, or kept by the client awaiting use. These substances are extremely susceptible to photo-oxidation: ergometrine [26], nifedipine [43], nitroprusside, riboflavin, and phenothiazines. When photochemical energy interacts with sensitive molecules, it produces intermediates called free radicals that can lead to further reactions. When exposed to high energy UV radiation, the majority of substances will break down as solutions. Antibiotics called fluoroquinolones have been proven to be photolytically cleavable [44].
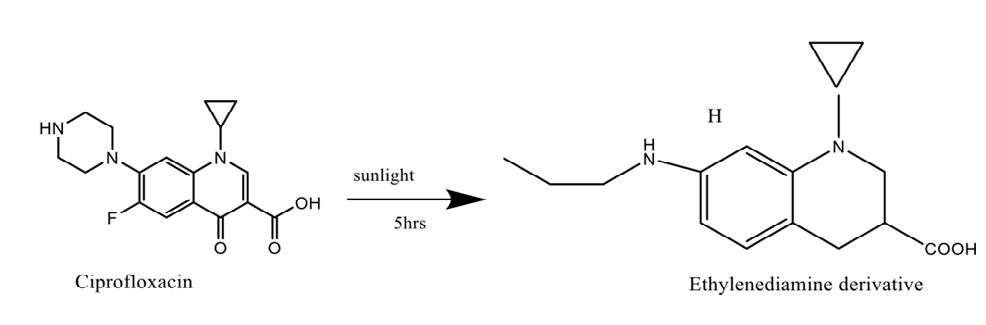
Decarboxylation: When heated, some dissolved carboxylic acids, such p-aminosalicylic acid, lose the carbon dioxide that was previously attached to the carboxyl group. Decarboxylation also took place when rufloxacin underwent a photoreaction [46].
Characterization of Impurities
Once an impurity has been identified, it must be estimated or quantified. Reference standard API is used to estimate contaminants in the new medication molecule initially. If analysis shows that an impurity present in the sample is higher than 0.1%, it must be labeled in accordance with FDA. guidelines. Even genuine samples with anticipated contaminants such as contaminants relating to raw materials, degradation products, Excipients, by-products, and process intermediates can all utilized to characterize impurities commonly both There are chromatographic and non-chromatographic methods. used to isolate contaminants before characterizing them. The following separation methods are most frequently used: Accelerated Solvent Extraction Method (ASE): The method utilized to expedite the extraction process is known as ASE. When compared to sonication or soxhlet extraction, ASE offers impurity extraction and is more faster. Increased pressure and temperature aid in the extraction process and make it more effective. Cost effectiveness is provided by a
90% reduction in solvent consumption. Supercritical Fluid Extraction (SFE): A substance acts as a supercritical fluid, which may diffuse through materials like a gas and dissolve solids like a liquid, when it is heated or compressed over its critical point. Utilizing supercritical fluid facilitates the extraction of extractant from matrix because mass transfer from sample to solvent occurs quickly and at a high extraction rate. Carbon Dioxide is the most favoured and efficient solvent for unstable chemicals due to its low critical temperature (31.1°C). High performance liquid chromatography (HPLC), gas chromatography, and supercritical fluid chromatography (SFC) can all be employed on an analytical level (GC). In contrast to HPLC, it can be utilized with non-volatile and thermally labile analytes, work with the universal flame ionization detector, and produce narrower peaks because of quick diffusion. Except in a few instances, such as chiral separations and the analysis of high- molecular-weight hydrocarbons, have the advantages of SFC been sufficient to replace the extensively used HPLC and GC in practice [47]. Column Chromatography: Based on the partition chromatography theory, column chromatography separates the components of the sample as it moves through the stationary phase while being influenced by the mobile phase. Many components elute out based on their affinity for the mobile phase. various rates from the column under gravity, which results in effective division. Sadly, the speed at which the solvent is very slow as it percolates through the column. The main benefit of column chromatography is that it can typically be used to sized for the current project. This is particularly beneficial if one attempting to separate and clean a reaction mixture as to pover in a series of reactions. The accompanying drawback is that column could take a while to correctly load. assemble and apply [48]. Thin Layer Chromatography (TLC): TLC is a method used to identify different components down to trace quantities. This method has been applied to create stability-indicating analytical method. Variability and non-quantify ability are its drawbacks. It is feasible to make the simplest, easiest decision at once. It can be utilized as a quantitative method, along with high performance thin layer with densitometric detection chromatography (HPTLC) for challenging substances to do an alternative chromatographic analysis because of the absence of chromophore. The basis for the TLC- based detection based on the interaction of the components and a tool for detection. During initial deterioration and stress tests, TLC is heavily utilised to count the amount of degradation products generated [49]. In comparison to traditional TLC, HPTLC is more sensitive and quicker. The advantages of HPTLC over TLC are I the need for a much smaller sample volume, (ii) the ability to measure more than 10 spots at once, and (iii) the ease with which different detectors; (iv) provide 3D images of every place, which are highly helpful for quantitative estimation; (v) have a shorter separation time than TLC. Utilizing the HPTLC approach, numerous stability indicating methods have been reported. tablets containing medications like telmisartan and ramipril [50], Prasugrel [51], Drotaverine and Aceclofenac in tablets [52], Pseudoephedrine and Cetirizine in pharmaceutical formulations [51]. Gas Chromatography (GC): Timing APIs is important for detecting contaminants, especially those that are volatile and thermo stable. It can be applied as a limit test for volatile contaminants like solvent residue in drug compounds. It is also used to characterize the starting ingredients for creating pharmacological compounds. Shorter run durations, increased sample throughput, less expensive columns, and a higher signal-to-noise ratio are a few benefits of GC. However, there are certain drawbacks, such as the need for careful attention when working on the instrument. Only situations in which the compounds may be vaporized without disintegrating and in which they can be evaporated at an acceptable temperature are appropriate for using gas chromatography (i.e. not so hot that it destroys the column packing). Only volatile or potentially volatile samples are used for analysis (reaction to form a volatile derivative). For the samples to remain intact when heated, they must be thermally stable. Once separated, it cannot be utilized to prepare samples for additional examination. The following issues could arise when injecting the sample: It is challenging to precisely quantify and inject such small samples (about 0.3 l) without the sample, for instance, evaporating. Sample loss could result from a leak in the rubber seal used to inject the sample. Small rubber septum fragments may adhere to the column and create “ghost peaks. In order to prevent vaporization, the sample might be directly injected into the heated portion of the injector. The quantitative accuracy and precision of GC can be on par with those of HPLC, especially when used in conjunction with an internal standard. Various articles of study focused on stability stating that there are available GC techniques, for example. identifying divalproex sodium contaminants in pharmaceutical formulations [52], fluconazole [53]. High Performance Liquid Chromatography (HPLC): HPLC is a versatile method of analysis as it is not limited to volatile or stable sample and separation is based on the fact that certain compounds have different migration rates on a particular stationary and mobile phase [54]. component separation using the HPLC technique and any suitable detector, such as a refractive index detector [55], PDA detector [56], An accurate, precise, and reliable method for quantitative analysis of pharmaceutical products and impurities is provided by fluorescence detectors, electrochemical detectors, electrical conductivity detectors, light scattering detectors, evaporative light scattering detectors, Corona Charged Aerosol Detector (CAD), Nano Quantity Aerosol Detector (NQAD), etc. HPLC also involves monitoring of stability of pure drug substance and in case of drug formulations. It can be used to measure degradation products, such as salicylic acid, betamethasone dipropionate, and their related chemicals in diprosalic lotion using a stability-indicating technique and HPLC [57], norfloxacin [58], allantoin [59]. Various Advantages of HPLC
- Speed
- High resolution
- Sensitivity
- Reproducibility of +/- 1%
- Accuracy
- Automation.
Various Disadvantages of HPLC
- Costy
- Complexity
- Low sensitivity for some compounds
- Irreversibly adsorbed compounds not detected
- Co-elution (two compounds escaping from the tubing at once) difficult to detect [60].
Flash Chromatography: Gravity fed chromatography is a slow and frequently ineffective method that can be replaced with flash chromatography. Flash chromatography is a mix of medium pressure and short column chromatography that is air pressure driven. The flow of the solvent is accelerated, thus reducing the amount of time required to purify the sample. Small silica gel particles (250–400 mesh size) are used in flash chromatography under pressure to push solvent across the stationary phase’s surface [45].
Capillary Electrophoresis (CE): According to their physical properties Charged molecules are separated by electrophoresis by migrating them in different directions based on their physical properties. Traditional electrophoresis has some drawbacks, including the requirement that the electrophoretic separation be completed before the molecules may be detected. To stop samples from being damaged by heat, voltages could be utilized. Both of these problems are helped by capillary electrophoresis issues as a result of the capillary tube’s high surface to volume ratio (due to a diameter of 25 to 100 m). It emits heat to deter samples from overheating and is employed to establish different drug-related contaminants that can be found in a variety of matrix examples [61]. When compared to reversed-phase (RP) HPLC, capillary electrophoresis has a distinct selectivity and is more versatile, with high separation efficiency, quick method development, and good resolution. By using CE, it is simple to separate impurities that are too polar in nature to provide enough retention with RP-HPLC. It has a reputation for being an effective tool for separating charged pharmaceuticals, contaminants, etc. There are numerous examples of this approach being utilized for impurity profiling in the literature, including the stability indicating capillary electrophoresis method for determining metformin hydrochloride in tablets [62]. It can also be coupled with a mass spectrometer to create a system called CE-MS using a variety of interfaces, such as a triple tube nebulizer [63, 64]. Electrically neutral analytes can now be separated using micellar electro kinetic chromatography (MEKC), a capillary electrophoresis (CE) separation technique. By incorporating an ionic micelle into the running solution, MEKC can be accomplished without changing the instrument, of CE. The separation tenet of it is based on the ionic micelles’ differential migration and under electrophoresis conditions, and on bulk running buffer interaction between the micelle and the analyte. Thus, MEKC’s Chromatography’s separation principle is similar. MEKC is an effective method, especially for the separation of tiny molecules, both charged and neutral, and produces high efficiency separation in a short length of time of sample and chemicals, such as the identification of fluticasone nasal sprays containing propionate by a validated stability-indicating for a pharmacological product used to decrease cholesterol, the MEKC method, MEKC stability indicating assay, and content uniformity determination were used [65, 66, 67]. Various Other Instrumental Methods are also used for the Purpose of Characterization of Impurities in Any Sample. These Methods include: Nuclear Magnetic Resonance (NMR) Spectroscopy: Nuclear magnetic resonance (NMR) spectroscopy is a necessary instrument for determining chemical structure due to its versatility. The main disadvantages of this approach are that it is very expensive, time-consuming, and takes a long time to analyse spectra. NMR can identify very fine structural components, works for both organic and inorganic molecules, and allows for both qualitative and quantitative evaluation. Advanced two- or multi-dimensional NMR spectroscopy, such as Correlation Spectroscopy (COSY), Nuclear Over hausser Enhancement Spectroscopy (NOESY), Hetero nuclear Correlation Spectroscopy (HETCOR), and Incredible Natural Abundance Double Quantum Experiment (INADEQUATE), are developed in addition to one dimensional NMR spectroscopy, which is used to study chemical bonding. realm of time The study of molecular dynamics in solutions uses NMR spectroscopy. In research studies, NMR frequencies between 60 MHz and 1 GHz have been applied. The measurement of chemical shifts in higher solution NMR spectroscopy is one of the main sources of chemical data. Numerous methods have been employed with various magnetic fields, including high-field superconducting magnets [68, 69, 70, 71, 72]. For instance, literature describes a stability suggesting proton NMR spectroscopic assay method for succinylcholine chloride injections [73].
Mass Spectroscopy (MS): MS has become an indispensable method for characterizing contaminants found in pharmaceutical products throughout the course of the last few decades. To enable direct interaction with separation techniques, interfaces with cutting-edge design and increased efficiency are being created. It can open up new possibilities for defining, quantifying, and monitoring drug- related chemicals in pharmaceutical APIs Formulations [74]. Mass spectroscopy has benefits like requiring a small sample size, being quick, differentiating isotopes, running mixtures with GC and LC, or running proteins in tandem. It can even provide elemental composition, but it doesn’t directly provide structural information (although we can frequently figure it out), requires pure compounds, and is challenging with non-volatile compounds. As the degree of conjugation increases, the spectra will shift towards the red area; for example, naphthalene absorbs light up to 300 nm and anthracene to roughly 400 nm. One of the simplest techniques for assessing the purity of a pharmacological ingredient is UV-VIS spectroscopy. Only samples having components or some of their derivatives that are spectrophotometrically active can be employed with UV- VIS spectroscopy [75]. UV-VIS spectroscopy is also used to characterize contaminants or degradation products created as a stability-indicating approach. Numerous techniques for analysing pharmacological substances utilizing UV-VIS spectroscopy as a stability indicator, such as tinidazole, are documented in the literature [76]. The method offers very high precision and accuracy while being very quick, easy, requiring little sample preparation, and cost-effective. It is non-destructive and effective for a wide range of compounds. On pure compounds, it can be applied quantitatively as well as qualitatively. Due to the addition of absorbance, UV has a limited application in mixture analysis. It needs specialized tools (such an ultraviolet light source and UV-transparent sample containers), and it is not selective for substances if they absorb at the same wavelength [77].
IR Spectroscopy: The sample is exposed to electromagnetic radiation with a wavelength between 500 cm-1 and 4000 cm-1, which affects the molecules’ bonds and causes them to stretch or bend as a result of the wavelength’s energy being absorbed. Wavelengths absorbed are indicative of different types of bonds, which aid in figuring out sample structure. IR spectroscopy can be used to characterize solid and semi- solid substances. In order to analyse medicine samples and identify the presence of contaminants in medications, IR spectroscopy creates a complicated but distinctive fingerprint of every molecule. It can also be used to identify the presence of drug polymorphs. Using photoacoustic spectroscopy in the infrared region, for instance, to evaluate 12-tungstophosphoric acid and similar salts, is a useful method for identifying contaminants in pharmaceutical products [78]. Compared to things like NMR, it is quick and inexpensive. Additionally, it is effective for a variety of materials and is much more effective at detecting chemicals than Raman spectroscopy or other comparable methods. The
techniques’ drawbacks include the time required for sample preparation and the fact that they can’t provide information as in-depth as others, like NMR. It is a destructive analytical approach, hence a non-destructive method like Raman should be used to analyse valuable or limited samples. There are many molecules that are not IR active and cannot be detected, therefore it is qualitative rather than quantitative.
Hyphenated Techniques: Hyphenated approaches use extremely complex instrumentation that consists of two or more devices combined with the aid of highly developed and effective interfaces. It is essential to use equipment like mass spectrometers, which are connected to a GC or HPLC, to identify small components including medicines, contaminants, degradation products, and metabolites in a variety of matrices. Numerous different chromatographic- spectroscopic configurations are available are ideal for determining contaminants at the beginning. For instance, preparative HPLC and NMR spectroscopy entail the extraction of bigger components using preparative HPLC and full structure elucidation through NMR spectroscopy. Although hyphenated techniques like HPLC-NMR or HPLC- MS are very effective, PDA detectors coupled to HPLC are a preferable choice due to their lower cost, lower operating costs, and lower time commitment. UVHPLC method to assess impurity profile can be used \sonly when contaminant is spectrophotometrically active [79]. Using LC and LC-MS/ MS techniques, a number of stability indicating approaches have been published in the literature. One such technique has been published for the assessment of contaminants in anastrozole tablets [80].
Applications of Isolation and Characterization of Impurities
A regulatory requirement for drug impurity profiling is to ensure the safety and effectiveness of pharmaceutical products. Drug monitoring and design have been the subject of numerous applications. Pharmaceutical chemicals’ quality, stability, and security, whether they are made synthetically or naturally either made using natural substances or recombinant techniques. Alkaloids, amines, amino acids, analgesics, antibiotics, anticonvulsants, antidepressants, sedatives, antineoplastic drugs, local anaesthetics, macromolecules, steroids, and other substances are used for various purposes [81].
Conclusion
A pharmaceutical ingredient must pass not only the CGMP, QC, QA, and water activity tests, but it must also meet the criteria for a new impurity. Separation and Characterization of impurities is necessary for gathering and assessing information that determines biological safety, highlighting the necessity and potential for drug impurity profiling in pharmaceutical research. A variety of instrumental analytical techniques have been regularly employed to separate and quantify the contaminants. In addition, there are several sources of organic impurities, including microbial contamination, API breakdown products, and traces of intermediates, making the identification and regulatory consideration of organic impurities a very difficult challenge. Even though ICH has a well-defined course of action in relation to contaminants, much more has to be done. Therefore, there is an urgent need for harmonized impurity requirements and standards.
References
-
Barber HJ (1978) Historical Aspects of Chemotherapy: Six Essays. May and Baker.
-
Ahuja S, Alsante KM (2003) Handbook of isolation and characterization of impurities in pharmaceuticals. 1st (Edn.), Academic Press, USA, Vol. 5, pp: 432.
-
Ahuja S (2006) Impurities Evaluation of Pharmaceuticals. Marcel Dekker, New York.
-
Ahuja S (2001) Handbook of modern pharmaceutical analysis. Academic press, USA, Vol 3, pp: 1-566.
-
Roy J (2002) Pharmaceutical impurities-A mini-review. AAPS Pharm Sci Tech 3(2): 1-8.
-
Ahuja S, Alsante KM (2003) Handbook of isolation and characterization of impurities in pharmaceuticals. 1st (Edn.), Academic press, USA, Vol. 5, pp: 432.
-
Keitel S (2006) Impurity Profiles in Active Pharmaceutical Ingredients. EU/Swissmedic GMP Workshop, Beijing University, China.
-
Alsante KM, Boutros P, Couturier MA, Friedmann RC, Harwood JW, et al. (2004) Pharmaceutical impurity identification: a case study using a multidisciplinary approach. Journal of Pharmaceutical Sciences 93(9): 2296-2309.
-
International Conference on Harmonization (2000) Draft Revised Guidance on Impurities in New Drug Substances. Federal Register Q3A(R) 65(140): 45085.
-
International Conference on Harmonization (2000) Draft Revised Guidance on Impurities In New Drug Products. Federal Register Q3B(R) 65(139): 44791.
-
International Conference on Harmonization (1997) Impurities, Q3C- Guidelines for Residual Solvents, Q3C. Federal Register 62(247): 67377-67388.
-
International Conference on Harmonization (1999) Specifications, Q6A: Test Procedures and Acceptance Criterial for New Drug Substances and New Drug Products. Chemical substances 65 (146): 67488.
-
Dhangar KR, Jagtap RB, Surana SJ, Shirkhedkar AA (2017) Impurity profiling of drugs towards safety and efficacy: theory and practice. Journal of the Chilean Chemical Society 62(2): 3543-3557.
-
Görög S, Babjak M, Balogh G, Brlik J, Csehi A, et al. (1997) Drug impurity profiling strategies. Talanta 44(9): 1517- 1526.
-
(2006) Impurities in new drug substances Q3A (R2). International conference on harmonization of technical requirements for registration of pharmaceuticals for human use, Geneva, Switzerland 25: 1-15.
-
Deshpande MM, Bhalerao MH, Pabale PD (2022) A Review on Impurity Profiling, Degradation Studies, and Bioanalytical Methods of Anti-diabetic Drugs. Journal of Pharmaceutical Research International 34(34B): 43-71.
-
(2014) Pharmacopoeia I. Ghaziabad: Indian Pharmacopoeial Commission. Ministry of Health and Family Welfare, Government of India.
-
Holm R, Elder DP (2006) Analytical advances in pharmaceutical impurity profiling. European Journal of Pharmaceutical Sciences 87: 118-135.
-
Beckett AH, Stanlake JB (2002) Practical Pharmaceutical Chemistry. 2nd (Edn.), CBS Publishers and distributors, London.
-
Gandhi SP, Dewani MG, Borole TC, Damle MC (2011) Development and Validation of Stability Indicating HPTLC Method for Determination of Ofloxacin and Ketorolac Tromethamine in Combination. Journal of Advanced Scientific Research 2(3): 77-82.
-
Connors KA, Amidon GL, Stella VJ (1986) Chemical stability of pharmaceuticals: a handbook for pharmacists. 2nd(Edn.), John Wiley & Sons, pp: 864.
-
Nagpal S, Upadhyay A, Bhardwaj TR, Thakkar A (2011) A review on need and importance of impurity profiling. Current Pharmaceutical Analysis 7(1): 62-70.
-
Görög S (2000) Identification and determination of impurities in drugs. 1st(Edn.), Elsevier, Budapest, Hungary Vol. 4, pp: 772.
-
Condorelli G, De Guidi G, Giulfrido S, Sortino S, Chillemi R, et al. (1999) Molecular mechanisms of photosensitization induced by drugs XII. Photochemistry and photosensitization of rufloxacin: An unusual photodegradation path for the antibacterials containing a fluoroquinolone-like chromophore. Photochem Photobiol 70(3): 280-296.
-
Hoerle SL, Evans KD, Snider BG (1992) HPLC Determination of Impurities in a 3rd Generation Cephalosporin. Eastern Analytical Symposium, New Jersey, Somerset.
-
Roy J, Bhuiyan K, Faruque A (1997) Injectable ergometrine: Stability and packaging for developing countries. Indian Drugs 34: 634-636.
-
Kumar V, Sunder N, Potdar A (1992) Critical factors in developing pharmnaceutical formulations-An overview Part 2. Pharm Technol 16: 86-88.
-
Smith A, Pennefather PM, Kaye SB, Hart CA (2001) Fluoroquinolones-place in ocular therapy. Drugs 61: 747-761.
-
Pilaniya K, Chandrawanshi HK, Pilaniya U, Manchandani P, Jain P, et al. (2010) Recent trends in the impurity profile of pharmaceuticals. Journal of Advanced Pharmaceutical Technology & Research 1(3): 302-310.
-
Riley TN (1998) Steric aspects of drug action. US Pharmacist 23(3): 40-51.
-
Hatakka H, Alatalo H, Palosaari S (2010) Effect of Impurities and additives on crystal Growth.
-
Jacobs P, Dewe W, Flament A, Gibella M, Ceccato A (2006) A new validation approach applied to the GC determination of impurities in organic solvents. Journal of pharmaceutical and biomedical analysis 40(2): 294- 304.
-
Gimeno P, Besacier F, Bottex M, Dujourdy L, Chaudron- Thozet H (2005) A study of impurities in intermediates and 3, 4 methylenedioxymethamphetamine (MDMA) samples produced via reductive amination routes. Forensic Sci Int 155(2-3): 141-57.
-
Markovic I (2007) Evaluation of safety and quality impact of extractable and leachable substances in therapeutic biologic protein products: a risk-based perspective. Informa 6(5): 487-491.
-
Roy J, Islam M, Khan AH, Das SC, Akhteruzzaman M, et al. (2001) Diclofenac sodium injection sterilized by autoclave and the occurrence of cyclic reaction producing a small amount of impurity. Journal of Pharmaceutical Sciences 90(5): 541-544.
-
Walker GJ, Hogerzeil HV (1988) Potency of ergometrine in tropical countries. Lancet 2(8607): 393.
-
Hogerzeil HV, Battersby A, Srdanovic V, Stjernstrom NE (1992) Stability of essential drugs during shipment to the tropics. BMJ British Medical Journal 304(6821): 210- 212.
-
Bari SB, Kadam BR, Jaiswal YS, Shirkhedkar AA (2007) Impurity profile: Significance in Active Pharmaceutical Ingredient. Eurasian journal of analytical chemistry 2(1): 1-32.
-
Hoq MM, Syeda M, Gomes DJ (1991) Development of appropriate preservative system for liquid antacid: bacterial contaminants in antacid samples. Bangladesh Journal of Microbiology 8(1): 5-9.
-
Buhler V (1988) Vademecum for Vitamin Formulation. 2nd(Edn.), Verl-Ges, Stuttgart, Germany, pp: 144.
-
Roy J, Mahmud M, Sobhan A, Aktheruzzaman M, Al- Farooque M, et al. (1994) Marketed vitamin B-complex injectables: stability and mutual interaction. Drug development and industrial pharmacy 20(13): 2157- 2163.
-
Hoerle SL, Evans KD, Snider BG (1992) HPLC Determination of Impurities in a 3rd Generation Cephalosporin. Eastern Analytical Symposium, New Jersey, Somerset.
-
Kumar V, Sunder N, Potdar A (1992) Critical Factors in Developing Pharmaceutical Formulations: an Overview. Pharmaceutical technology 16(4).
-
Bari SB, Kadam BR, Jaiswal YS, Shirkhedkar AA (2007) Impurity profile: Significance in Active Pharmaceutical Ingredient. Eurasian journal of analytical chemistry 2(1): 32-52.
-
Still WC, Kahn M, Mitra A (1978) Rapid chromatographic technique for preparative separations with moderate resolution. J Org Chem 43(14): 2923-2925.
-
Bart CJ (2005) Separation Techniques - Additives in Polymers: indusrtrial analysis and applications. John Wiley and Sons, USA, pp: 212.
-
Bakshi M Singh S (2002) Development of validated stability-indicating assay methods-critical review. Journal of pharmaceutical and biomedical analysis 28(6):1011-1040.
-
Pal K, Raju MB, Kumar KP, Chakravorthy R (2010) Simultaneous Determination of Telmisartan and Ramipril in Tablet Dosage Form by Spectrophotometry. Parameters 97(101.60): 97-103.
-
Borole TC, Mehendre R, Damle MC, Bothara KG (2010) Development and validation of stability indicating HPTLC method for determination of Prasugrel. J Chem Pharm Res 2(4): 907-913.
-
Wankhede SB, Mahale DK, Chitlange SS (2010) Stability-indicating HPTLC method for simultaneous determinationof Drotaverine and Aceclofenac in tablet formulation. Der Pharma Chemica 2(5): 107-117.
-
Makhija SN, Vavia PR (2001) Stability indicating HPTLC method for the simultaneous determination of pseudoephedrine and cetirizine in pharmaceutical formulations. J Pharm Biomed Anal 25: 663-667.
-
Subasranjan A, Suresh P, Srinivasulu C, Hemant R (2010) A validated stability-indicating gas chromatography method for determination of divalproex sodium impurities in pharmaceutical preparation. Drug Test Anal 2(4): 182-187
-
Hunt-Fugate AK, Hennessey CK, Kazarian CM (1993) Stability of fluconazole in injectable solutions. American J Hospital Pharm 50(6): 1186-1187.
-
Pal K, Raju MB, Kumar KP, Chakravorthy R (2010) Simultaneous Determination of Telmisartan and Ramipril in Tablet Dosage Form by Spectrophotometry. Research Journal of Pharmacy and Technology 3(3): 97- 103.
-
Raymond S (1995) Chromatographic detectors design: Function and operation. Chromatographic science series 73: 201-204.
-
Ankush P, Tejashree R, Vishal S, Ravindra T(2020) Recent Approaches for Impurity Profiling: A Review. International Journal for Research Trends and Innovation 5(5): 1-5.
-
Shou M, Galinada WA, Wei YC, Tang Q, Robert J, et al. (2009) Development and validation of a stability- indicating HPLC method for simultaneous determination of salicylic acid, betamethasone dipropionate and their related compounds in Diprosalic Lotion. Journal of pharmaceutical and biomedical analysis 50(3): 356-361.
-
Rao RN, Nagaraju V (2004) Separation and determination of synthetic impurities of norfloxacin by reversed-phase high performance liquid chromatography. Journal of pharmaceutical and biomedical analysis 34(5): 1049- 1056.
-
Zaidi ZR, Sena FJ, Basilio CP (1982) Stability assay of allantoin in lotions and creams by high-pressure liquid chromatography. Journal of Pharmaceutical Sciences 71(9): 997-999.
-
Xiang Y, Liu Y, Lee ML (2006) Ultrahigh pressure liquid chromatography using elevated temperature. Journal of Chromatography A 1104(1-2): 198-202.
-
Nagpal S, Upadhyay A, Bhardwaj T, Thakkar A (2011) A review on need and importance of impurity profiling. Current Pharmaceutical Analysis 7(1): 62-70.
-
Hamdan II, Jaber AB, Abushoffa AM (2010) Development and validation of a stability indicating capillary electrophoresis method for the determination of metformin hydrochloride in tablets. Journal of pharmaceutical and biomedical analysis 53(5): 1254- 1247.
-
Sauber C, Ross G Impurity Profiling with Capillary Electrophoresis/Ion Trap Mass Spectrometry. Application Note, Agilent Technologies, USA.
-
Blomberg LG, Wan H (2000) Determination of Enantiomeric Excess By Capillary Electrophoresis. Electrophoresis: An International Journal 21(10): 1940- 1952.
-
Terabe S (2009) Capillary separation: micellar electrokinetic chromatography. Annual Review of Analytical Chemistry 2: 99-120.
-
Silva SSM, Silva MDL, Avila FB, Dalmora SL (2010) Determination of fluticasone propionate in nasal sprays by a validated stability-indicating MEKC method. Journal of chromatographic science 48(8): 641-646.
-
Bretnall AE, Hodgkinson MM, Clarke GS(1997) Micellar electrokinetic chromatography stability indicating assay and content uniformity determination for a cholesterol- lowering drug product. Journal of pharmaceutical and biomedical analysis 15(8): 1071-1075.
-
Hobbie RK (1988) Intermediate Physics for Medicine and Biology. Springer, New York.
-
Smalley RE (1997) Discovering the fullerenes. Reviews of Modern Physics 69(3): 723.
-
Spiess HW (1978) Rotation of molecules and nuclear spin relaxation. Dynamic NMR spectroscopy Springer, Berlin, Heidelberg, pp: 55-214.
-
Pavia DL, Lampman GM, Kriz GS (2009) Introduction to Spectroscopy. Thomson Learning Inc Vol. 3, pp: 540.
-
Silverstein RM, Bassler GC (1962) Spectrometric identification of organic compounds. Journal of Chemical Education 39(11): 546.
-
Hanna GM, Lau-cam CA (1985) A stability indicating proton NMR spectroscopic assay method for succinylcholine chloride injections. Analytical Letters 18(17): 2183-2194.
-
Fiori J, Bragieri M, Zanotti MC, Liverani A, Borzatta V, et al. (2005) Liquid chromatography–tandem mass spectrometry for the identification of impurities in dallethrin samples. Journal of Chromatography A 1099(1-2):149-156.
-
Greenlief (2004) UV Vis Absorption Spectroscopy. CH 4200, Fall.
-
Salo JP, Salomies H(2003) Two stability-indicating UV spectrophotometric methods for the analysis of hydrolyzed tinidazole. Journal of pharmaceutical and biomedical analysis 31(3): 523-536.
-
Highfield JG, Moffat JB (1984) Characterization of 12-tungstophosphoric acid and related salts using photoacoustic spectroscopy in the infrared region: I Thermal stability and interactions with ammonia. Journal of Catalysis 88(1): 177-187.
-
Skoog DA, Holler FJ, Crouch SR (2007) Principles of instrumental analysis. Thomson brooks, Cole, Canada.
-
Berridge JC (1995) Impurities in drug substances and drug products: new approaches to quantification and qualification. Journal of pharmaceutical and biomedical analysis 14(1-2): 7-12.
-
Reddy YR, Nandan SR, Bharathi DV, Nagaraju B, Reddy SS, et al. (2009) LC and LC–MS/MS study of forced decomposition behavior of anastrozole and establishment of validated stability-indicating analytical method for impurities estimation in low dose anastrozole tablets. Journal of pharmaceutical and biomedical analysis 50(3): 397-404.
-
Gupta R, Jain S, Gupta A (2014) A review on the impurity profile of pharmaceuticals. International journal of drug formulation and research.
- Spectrophotometric Determination of Lanthanum (III) and Some Rare Earths with Xylenol Orange
- Introduction and Sources of Molluscicides
- Trimetazidine: An Antianginal Drug and Not Only!
- Nature Inspired Discovery and Development of Antibacterials: An Update
- Fungal Biodegradation of Polycyclic Aromatic Hydrocarbons (PAHs)
- Next Generation Tools in mRNA Purification: The Role of Continuous Raman Spectroscopy Testing with Pretreatment of the Sample