Mechanisms of RNA Interference-Based Antiviral Strategies in Mammals
Mammalian species possess sophisticated innate immune mechanisms that collectively combat a wide array of viral pathogens, with the interferon (IFN) response being extensively researched; however, the emerging role of antiviral RNAi in mammals is garnering significant interest, as evidenced by research indicating that Dicer, an enzyme essential for processing dsRNA, demonstrates reduced activity in vitro and that the IFN response may overshadow or inhibit the antiviral functions of RNAi in mammalian cells, raising questions about the functional relevance of RNAi in antiviral defense within mammalian somatic cells. This complexity is further illustrated by evidence suggesting a mutual inhibition between RNAi and the IFN pathway, where proteins like LGP2 inhibit Dicer, thus affecting the enzyme's capacity to process long dsRNA versus precursor miRNAs, potentially influencing antiviral efficacy. While traditional protein-guided immune responses are critical for survival in viral environments, small RNA-mediated antiviral systems utilizing complementary base pairing to silence non-self genetic material also play a crucial role, with emerging data indicating that miRNAs, siRNAs, piRNAs, and tRNAs can directly target virus-derived nucleic acids. This review aims to highlight some of the recent progress in understanding mammalian antiviral RNAi mechanisms.
Abbreviations
aviD: Antiviral Dicer; AGO2: Argonaute Protein 2; AGO: Argonaute Protein; dsRNA: Double-stranded RNA; dsRNAi: Double-stranded RNA interference; vRI-dsRNA: Double- stranded RNA Viral Replicative Intermediates; hDcr: Human Dicer; HIF: hypoxia-inducible Factor; IFN: Interferon; ISG: Interferon-stimulated Gene; IFNAR: Interferon-α/β Receptor; Lgr5: Leucine-rich Repeat-containing G Protein- coupled Receptor 5; LGP2: Laboratory of Genetics and Physiology 2; MAVS: Mitochondrial Antiviral Signaling Protein; PACT - Protein Activator of PKR; PAZ: Piwi Argonaute Zwille; piRNAs: PIWI-interacting RNAs; miRNAs: precursor microRNAs; pri-miRNAs: Primary miRNAs; PACT: Protein Activator of PKR; RNase III: Ribonuclease III; RLR: RIG-I- like Receptor; RNAi: RNA Interference; RISC: RNA-induced Silencing Complex; ssRNA: Single-stranded RNA; siRNAs: Small Interfering RNAs; TRBP: Trans-activation Response RNA-binding Protein; tRNAs: Transfer RNAs; vRNA: Viral RNA; VSR: Viral Suppressors of RNA Interference; vsiRNA: Virus-derived small interfering RNA.
Introduction
RNAi has emerged as one of the most promising biological mechanisms in antiviral defense across a wide array of organisms, including mammals. This cellular process, which involves the sequence-specific degradation of RNA molecules, plays a critical role in regulating gene expression and maintaining cellular homeostasis. The RNAi
pathway discovery has paved the way for exploring RNAi- based antiviral strategies, which leverage the cell’s intrinsic machinery to combat viral infections. These strategies involve the use of siRNAs or miRNAs that can specifically bind to and degrade viral RNA, thus inhibiting viral replication and spread. The ability to target specific RNA sequences makes RNAi an attractive tool for developing therapies against a wide range of viruses, including those that currently have no effective treatments. The significance of RNAi-based antiviral strategies in human health cannot be overstated, especially in light of the growing global health threat posed by emerging and re-emerging viral diseases. Viral infections remain significant challenges to modern medicine due to their rapid mutation rates, which often render conventional treatments ineffective. RNAi-based strategies offer the potential to complement existing antiviral measures by providing a flexible, targeted approach that can be rapidly developed in response to new viral threats.
In addition to combating viral infections in humans, RNAi has potential applications in the treatment of viral diseases in livestock, which can have devastating economic impacts. Traditional measures to control such outbreaks often involve mass culling, which is both costly and ethically challenging. RNAi-based antiviral approaches could provide a more humane and effective solution by preventing viral replication at the molecular level, thus controlling the spread of the virus without the need for drastic interventions. RNAi-based antiviral therapies could address the growing issue of drug- resistant viruses. Antiviral drug resistance is a significant problem, particularly in the case of HIV and hepatitis C virus (HCV), where long-term treatment with antiviral drugs can lead to the emergence of resistant viral strains. The ability of RNAi to target highly conserved regions of viral genomes offers a potential solution to this issue, as it may be more difficult for viruses to develop resistance to RNAi-based therapies than to traditional drugs. This characteristic makes RNAi a valuable tool in the ongoing fight against viral diseases that evolve and adapt to evade conventional treatments. The development of RNAi-based antiviral strategies is not without challenges. Effective delivery of RNAi molecules to target tissues, ensuring stability and avoiding off-target effects, remains a significant hurdle in therapeutic applications. This article highlights some of the key mechanisms involved in the RNAi pathways of mammals.
An Overview about RNAi Pathway
RNAi is a process that leads to the silencing of gene expression after transcription, a phenomenon initially noticed in pigmented petunia flowers in 1990 [1]. This gene-silencing effect was found to be induced by double-stranded RNA (dsRNA), as demonstrated by Fire et al.,1998 through their work with the nematode Caenorhabditis elegans [2, 3]. Their discovery revealed that the presence of dsRNA significantly decreased the levels of corresponding mRNA, thus effectively silencing the gene [4]. The RNAi pathway is controlled by Dicer, an enzyme belonging to the RNase III family, which plays a critical role in the generation of small RNA molecules from longer RNA precursors (Figure 1). Dicer processes these long dsRNA molecules into siRNAs and converts pre-miRNAs into mature miRNAs by cleaving hairpin structures [5]. These small RNA fragments are then incorporated into the RISC, where they guide the AGO to recognize and bind to complementary mRNA sequences [6]. Argonaute, an essential component of RISC, facilitates the degradation or translational repression of the target mRNA, thereby silencing the gene.
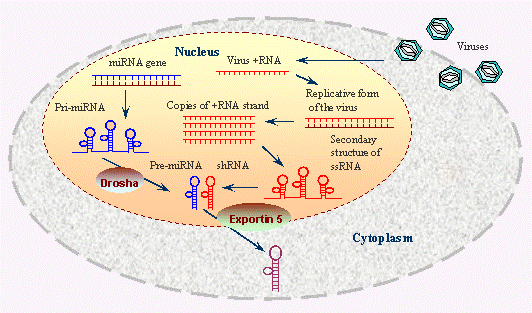
Figure 1: The processes occurring within the nucleus and cytoplasm of a cell during both miRNA biogenesis and viral replication. Within the nucleus, miRNA genes are transcribed into primary pri0-miRNAs, which are then processed by the Drosha enzyme into pre-miRNAs. These pre-miRNAs are exported to the cytoplasm via Exportin 5, where they are further processed into mature miRNAs that can regulate gene expression. The nucleus is shown hosting viral replication, where vRNA is transcribed and replicated, forming dsRNA intermediates and ssRNA that can form secondary structures. These viral RNAs can be shuttled to the cytoplasm to continue the infection cycle. Image Source: https://www.ncbi.nlm.nih.gov/probe/docs/techrnai/
Progress in Mammalian Antiviral RNAi Research
Multiple studies have highlighted that RNAi serves as a well-preserved antiviral defense mechanism across a diverse array of eukaryotic species, including mammals (Figure 2) [2]. RNAi operates through the sequence-specific degradation of RNA, which is mediated by small RNA molecules such as siRNAs and miRNAs. These small RNAs guide the RISC to target and degrade complementary viral RNA, thereby mitigating viral infections in these organisms. In contrast, the presence and functionality of antiviral RNAi in mammals have been subjects of ongoing debate. This debate arises primarily from the existence of alternative antiviral defense mechanisms in mammals, such as the interferon (IFN) response and the adaptive immune system, which are not present in plants and invertebrates. One perspective suggests that antiviral RNAi and the IFN system may engage in mutual inhibition. Specifically, the IFN response can interfere with the RISC complex—a critical component of the RNAi machinery—thereby disrupting its ability to process and degrade viral RNAs. The protein LGP2, which plays a crucial role in both the IFN response and adaptive immunity, has been shown to inhibit the processing of precursor miRNAs and long dsRNA by the Dicer enzyme. This inhibition could further compromise the effectiveness of the RNAi pathway in mammals [7, 8]. Conversely, some researchers propose that a conflict exists between these antiviral systems in mammals. This viewpoint is based on observations that, although vsiRNAs are produced in response to viral infections, they do not appear to significantly reduce viral replication. This lack of effective antiviral response has led to the hypothesis that the RNAi pathway is functionally compromised or non- functional in mammals, suggesting that other mechanisms, such as the robust IFN response and adaptive immunity, might dominate in providing antiviral defense [9, 10].
![Figure 2: The dual mechanisms by which mammalian cells restrict viral infections: the IFN response and antiviral RNAi. On the left, the IFN response is depicted, where viral dsRNA is detected by RLRs such as LGP2, RIG-I, and MDA5, leading to the activation of type I and III interferons (IFN-α, IFN-λ, IFN-β). These interferons then induce the expression of ISGs that collectively inhibit viral replication. On the right, the antiviral RNAi pathway is shown, starting with the processing of viral dsRNA by Dicer into 22-nucleotide viRNAs, which are then incorporated into the RISC. The RISC, guided by viRNAs, cleaves the target viral RNA, leading to the degradation of viral RNA and the inhibition of viral replication. Image Source: [17].](/fulltextimages/13258/fig_2.png)
Figure 2: The dual mechanisms by which mammalian cells restrict viral infections: the IFN response and antiviral RNAi. On the left, the IFN response is depicted, where viral dsRNA is detected by RLRs such as LGP2, RIG-I, and MDA5, leading to the activation of type I and III interferons (IFN-α, IFN-λ, IFN-β). These interferons then induce the expression of ISGs that collectively inhibit viral replication. On the right, the antiviral RNAi pathway is shown, starting with the processing of viral dsRNA by Dicer into 22-nucleotide viRNAs, which are then incorporated into the RISC. The RISC, guided by viRNAs, cleaves the target viral RNA, leading to the degradation of viral RNA and the inhibition of viral replication. Image Source: [17].
However multiple studies have confirmed the presence of antiviral RNAi in both undifferentiated and differentiated mammalian cells [2, 11, 12, 13, 14]. dsRNA-binding proteins have been seen to have a reciprocal role in controlling the IFN system and antiviral RNAi. For instance, during infections like those brought on by the virus like the Sendai virus, the trans- activation response RNA-binding protein (TRBP) interacts with LGP2 to affect IFN production in addition to increasing the cleavage activity of Dicer on pre-miRNA [15, 16].
Mammalian Dicer's Inefficiency in Processing Long dsRNA
The molecular properties of Dicer, a critical enzyme in the dsRNAi pathway, significantly influence its effectiveness in mammalian cells. Dicer is a multi-domain enzyme characterized by a complex structural arrangement, which includes several key functional domains. At its N-terminus, Dicer contains an ATPase site within the DExD/H helicase domain, which is essential for unwinding RNA duplexes. This is followed by two tandem RNAse III domains, which are responsible for cleaving each strand of the dsRNA substrate. The enzyme also features a PAZ domain, which binds to the 3′ 2-nucleotide overhangs at the ends of dsRNA substrates, facilitating the recognition and binding of the RNA. Dicer possesses a domain of unknown function (DUF283) and a C-terminal double-stranded RNA-binding domain, which further contribute to its substrate specificity and catalytic activity [17]. Despite its critical role, hDcr exhibits different efficiencies in processing dsRNA compared to pre-miRNA. In vitro studies have shown that hDcr is less efficient at processing long dsRNA into siRNAs than it is at processing precursor miRNA into mature miRNAs [18, 19]. This differential efficiency suggests that the molecular structure of Dicer may impact its substrate preferences and processing capabilities.
Interestingly, modifying the Dicer enzyme can alter its activity. For example, deleting or partially proteolyzing the helicase domain of Dicer results in an increased rate of dsRNA cleavage, although this modification has only a modest effect on pre-miRNA processing [18]. This indicates that the helicase domain may play a role in modulating the enzyme’s activity towards dsRNA substrates. A deletion mutant of hDcr, which lacks nearly the entire helicase domain, demonstrates enhanced ability to process long dsRNA and long hairpin RNAs into siRNAs. This modification has enabled effective dsRNAi activity in engineered cells, suggesting that removing the helicase domain can improve the enzyme’s efficiency for specific RNA substrates [20]. In mouse oocytes, which are known to actively engage in dsRNAi, a truncated isoform of Dicer known as DicerO—lacking the N-terminal helicase domain—efficiently processes both endogenous and ectopically expressed long hairpin RNAs into siRNAs [21]. This observation highlights that in certain cellular contexts, such as mouse germ cells, alternative forms of Dicer with modified domain structures can facilitate effective RNA processing.
On the other hand, the expression of DicerO outside of mouse germ cells has not been detected, and in humans, and truncated Dicer isoforms have only been observed in specific cancer cell lines [22, 23]. These findings suggest that while the helicase domain may impose limitations on Dicer’s catalytic activity for long dsRNA, its regulation and alternative splicing might be cell type-specific and context- dependent. This accentuates the complexity of Dicer’s role in RNAi and its potential for diverse functional outcomes depending on its structural configuration and the cellular environment. According to structural research, co-factors like PACT and TRBP can cause the Dicer helicase domain to shift conformation, potentially simulating the effects of the domain’s deletion [23, 24]. This conformational change facilitated by TRBP and PACT suggests they play a role in modulating Dicer’s ability to process long dsRNA in vivo [19, 25]. During replication, RNA viruses produce vRI-dsRNA, which are cleaved by Dicer to generate 21–23 nucleotide vsiRNAs with 2-nucleotide 3’ overhangs [2]. These vsiRNAs then enter AGO2, the sole AGO protein in mammals with slicing activity, to play a downstream role in antiviral immunity [26, 27]. The discovery of an isoform of Dicer, termed aviD, which lacks exons 7 and 8, resulting in the absence of the Hel2i subdomain was seen to have enhanced antiviral RNAi capability and protects stem cells from Zika virus (ZIKV) and severe acute respiratory syndrome coronavirus 2 (SARS- CoV-2) infections by processing viral dsRNAs into siRNAs more effectively [28, 29].
IFN Response vs. dsRNAi
The antagonistic interaction between the IFN system and dsRNAi was investigated in somatic cells that were genetically engineered to lack IFNAR or MAVS in a work carried out by Maillard, et al. [30]. Injecting dsRNA into the cells was reported to have resulted in sequence-specific gene silencing requiring Ago2, which resulted in the accumulation of siRNAs in a Dicer-dependent manner [30]. Additional investigation conducted in 2018 by Van der Veen et al. revealed that the IFN system actively inhibits dsRNAi by triggering the expression of LGP2 [31]. This protein binds to Dicer and prevents lengthy dsRNA from being processed into siRNAs. Interestingly, despite the fact that LGP2 is also known to interact with TRBP, a co-factor of Dicer, and limit the processing of certain TRBP-bound miRNAs [32, 33], the binding of LGP2 to Dicer in this investigation did not influence the production of two housekeeping miRNAs. It remains to be clarified whether LGP2 further inhibits dsRNAi through its interaction with TRBP.
The rationale behind the inhibition of dsRNAi in somatic cells during an IFN response is not fully understood. Insights might be drawn from Girardi et al., 2015, who observed that mammalian cells stably expressing Drosophila dcr- 2 to artificially enhance dsRNAi exhibited a reduced IFN response upon treatment with poly(I:C), a synthetic dsRNA analog [34]. Both viral infection and poly(I:C) treatment have been shown to induce poly-ADP-ribosylation of Ago2 and other components of the RISC, which obstructs RISC activity and thereby alleviates miRNA-mediated repression of some interferon-stimulated genes (ISGs) [35]. This suggests that inhibiting Dicer and RISC might be crucial for the effective activation of the IFN pathway, potentially by preserving dsRNA substrates necessary for RLR activation [17]. Moreover, maintaining dsRNA in infected cells safeguard against the disruption of function of antiviral proteins encoded by ISGs. For instance, the protein kinase PKR involves dsRNA longer than 30 nucleotides to dimerize and become active, leading to translational repression [36]. If Dicer cleaves long dsRNA, it might deprive the cell of necessary substrates for PKR activation or cause an accumulation of 21-22 nucleotide siRNA duplexes that could bind to PKR monomers and prevent their dimerization, thereby blocking PKR activation [17].
During the coevolution of viruses and their hosts, a range of VSRs have emerged, developed by various viruses to counteract the host’s antiviral responses and enhance their ability to invade. These VSRs include proteins such as NoV B2 from Nodamura virus, NS2A from Dengue virus 2, Semliki Forest virus (SFV) capsid protein, and Rubella Virus capsid protein [2, 37, 38]. Notably, among these suppressors, NoV B2 and DENV2 NS2A are unique in their ability to function as dsRNA-binding proteins with IFN-independent VSR activity. In contrast, other VSRs, such as Influenza A virus NS1 (IAV NS1), exhibit a dual role by interacting with dsRNA to both suppress RNA interference and simultaneously antagonize the interferon response [39]. Lately, Fang et al., 2021 had designed VSR-targeting peptides (VTPs) that disrupt the function of human enterovirus 71 (HEV71) 3A, thereby unleashing the antiviral RNAi response to reduce viral replication, highlighting a promising therapeutic strategy [40]. It has gained significant attention in the field of gene regulation, particularly in its application to cancer, viral infections, cardiovascular diseases, diabetes, and stem cell therapy. Cancer-related oncogenes, tumor suppressor genes, and regulatory genes are prominent targets of RNAi therapy. By selectively silencing cancer-associated genes, RNAi can inhibit tumor growth, angiogenesis, and metastasis. For instance, silencing Lgr5, a gastric cancer marker, reduces angiogenesis, and siRNA targeting HIF-1α inhibits osteosarcoma angiogenesis [41, 42]. RNAi has shown potential in overcoming drug resistance to VEGF inhibitors by targeting VEGF in combination with bevacizumab, extending the efficacy of treatment [41].
In the context of viral infections, RNAi offers an innovative alternative to traditional antiviral therapies by silencing key viral or host genes involved in viral replication. Studies have demonstrated effective inhibition of viral replication in HIV and respiratory syncytial virus using siRNA and miRNA [41]. RNAi targeting hepatitis B and C viruses (HBV, HCV) has shown promise in reducing viral load, stimulating immune responses, and improving treatment outcomes. In particular, targeting miR-122 in HCV with miRNA inhibitors like Miravirsen has shown success in clinical trials [43]. RNAi-based miRNA therapeutics have also been explored for conditions like hypertension and atherosclerosis. Similarly, miRNA deregulation has been linked to diabetes, influencing insulin secretion and β-cell differentiation. RNAi- based approaches have demonstrated potential in β-cell repair and the treatment of diabetes related problems [44]. Lastly, RNAi can enhance stem cell therapy by regulating stem cell migration, inhibiting fibrosis, and modulating stem cell-derived factors [41]. This highlights the broad therapeutic potential of RNAi across various diseases. Despite the significant progress in understanding antiviral RNAi, several fundamental questions remain. These include the inconsistent detection of vsiRNAs, the efficiency of full- length Dicer cleavage, and the failure to observe increased viral replication in Dicer-deficient mammalian cells [45]. Addressing these questions is crucial for advancing research on antiviral RNAi.
Absence of Stable Mammalian vsiRNAs Detection During Viral Infection
In mammals, the production of vsiRNAs is a key marker of the antiviral RNAi response during viral infections. Studies have shown that vsiRNAs can be generated from viral genomes using mutant viruses that are deficient in VSRs. For example, NS1-deficient influenza A virus (IAV) have been shown to produce vsiRNAs when tested in mammalian cells [2, 46]. These vsiRNAs are generated as part of the host’s antiviral response, aiming to inhibit viral replication by degrading viral RNA. Moreover, vsiRNAs have been identified in experiments involving viruses in various cell types, including both undifferentiated and somatic cells [47]. This indicates that the production of vsiRNAs is not restricted to genetically modified viral strains but can also occur with naturally occurring viruses under specific conditions.
However, despite these findings, several studies have not detected vsiRNAs when analyzing a range of wild-type viruses. For instance, deep sequencing efforts have failed to identify vsiRNAs for negative-stranded viruses like Vesicular Stomatitis Virus (VSV) and IAV, as well as for positive-stranded viruses such as Poliovirus (PV), Hepatitis C virus (HCV), Dengue virus (DENV), Sindbis virus (SINV), coxsackievirus B3 (CVB3), and human enterovirus 71 (HEV71) across various mammalian cell lines [2, 48]. This lack of detection might be attributed to several factors. One possibility is that vsiRNAs are present at very low abundance within the total RNA pool, making them difficult to detect using standard sequencing techniques. The expected vsiRNA characteristics, such as the typical 22-nucleotide length, might not always be present or may be present in such low quantities that they fall below the detection threshold of deep sequencing technologies [45]. The absence of detectable vsiRNAs in these studies could also be due to differences in the mechanisms of RNAi between various viral systems or cell types. Some viruses may evade or suppress the RNAi response more effectively, or the host cells may not mount a robust RNAi response under certain conditions. These observations emphasize the need for further investigation into the conditions and mechanisms that govern vsiRNA production and the antiviral RNAi response in mammals, as well as the potential factors that might influence the detection and efficacy of these small RNA molecules in combating viral infections.
Several factors may account for the challenges encountered in detecting vsiRNAs in mammalian cells infected with various viruses. One potential issue is that earlier research may not have employed deep sequencing techniques with sufficient depth or sensitivity to detect vsiRNAs reliably. Deep sequencing is crucial for identifying low-abundance RNA species, such as vsiRNAs, and inadequate sequencing depth could result in the failure to capture these small RNA molecules effectively [49]. Another significant factor could be the interference of RNase L cleavage products with the detection of vsiRNAs. RNase L, an enzyme activated during antiviral responses, cleaves RNA molecules, including viral and host RNAs, into smaller fragments. This cleavage can generate RNA products that overlap with the size range of vsiRNAs, potentially obscuring the characteristic 22-nucleotide peak associated with vsiRNAs in sequencing data [2]. For instance, Girardi et al., 2013 investigated Sindbis virus (SINV)-infected HEK293 and Vero cells and proposed that some viral small RNAs observed were actually degradation products resulting from RNase L activity, rather than genuine vsiRNAs [50]. Moreover, differences in cell lines used for vsiRNA detection could contribute to variability in results. In particular, the RNase L pathway might be overly activated in ex vivo conditions, such as those used in earlier studies, leading to excessive RNA degradation and potential misinterpretation of RNA profiles [50]. Conversely, in vivo conditions may provide a more regulated environment where RNase L activity is controlled more precisely. This controlled regulation might facilitate the clearer detection of vsiRNAs, as observed in studies involving viruses like Zika virus (ZIKV) and Sindbis virus (SINV) where vsiRNAs were successfully identified [47].
Full-Length Dicer also Processes dsRNA
Insects possess two distinct Dicer proteins, Dicer-1 and Dicer-2, where Dicer-1 is involved in miRNA production, and Dicer-2 is responsible for generating siRNAs [16]. For a long time, it was believed that mammals only had a single type of Dicer that could process both miRNAs and siRNAs [2]. However, an isoform of Dicer distinct from the full-length Dicer was discovered in mouse oocytes by Flemr, et al. due to a loss in the N-terminal helicase domain [21]. When compared to the full-length Dicer, this isoform showed improved effectiveness in cleaving long hairpin dsRNA substrates, suggesting that these oocytes may have a higher capacity for antiviral RNAi [21]. Given this, Kennedy, et al. created a mutant form of human Dicer that does not have the amino- terminal helicase domain (N1 hDcr), and they produced it in NoDice/ΔPKR cells together with 257-bp dsRNA and empty vector, wild-type hDcr, or N1 hDcr [51]. Their results demonstrated that expressing N1 hDcr significantly increased the production of short RNA reads from 257-bp dsRNA from 0.25% to 23.9%, compared to NoDice/ΔPKR cells expressing the empty vector. In contrast, expressing wild-type hDcr increased short RNA reads to 7.04% [51]. Despite the fact that the N1 hDcr mutant generated 3.39 times more short RNA reads than wild-type hDcr, the wild-type hDcr was still capable of processing long dsRNA into siRNAs. The findings of Wang, J. and Li, Y. (2024) align with this, showing that Dicer efficiently processes IAV-derived dsRNA into vsiRNAs, even when IFN is activated by IAV infection in mammalian somatic cells [2, 52]. It’s interesting to note that Poirier et al., 2021 compared the in vitro cleavage efficiency of synthetic dsRNA substrates by aviD and full-length Dicer and discovered that, presumably, aviD still possessed some dicing activity because its cleavage efficiency was roughly twice that of full-length Dicer [28]. They noted that aviD showed increased resilience to LGP2 inhibition [28]. Future studies should investigate whether Dicer’s capacity to cleave dsRNA in vivo is influenced by other cofactors, such as PKR- associated activator (PACT), protein kinase RNA-activated (PKR), and adenosine deaminases acting on RNA 1 (ADAR1) [53, 54].
The significant role of antiviral RNAi in managing viral infections in mammals is accentuated by the theoretical consequences of depleting Dicer, a key protein in this defense mechanism. If Dicer, which is essential for antiviral RNAi, is knocked down in mammalian cells, it is expected that viral replication would increase. This hypothesis finds support in the work of Xu, et al. who documented enhanced Zika virus replication in human neural progenitor cells with reduced Dicer levels [12]. In contrast, Cullen, et al. did not observe a corresponding increase in viral replication when Dicer-knockout human somatic cells were exposed to various viruses [55]. Findings from Witteveldt, et al. showed that mouse embryonic stem cells lacking Dicer displayed heightened resistance to several viral infections [56]. Further research has demonstrated that mammalian cells deficient in Dicer accumulate endogenous dsRNAs, including Alu RNAs—predominantly found in the human genome—and B2 RNAs—prevalent in the mouse genome [57]. Typically, in healthy mammalian cells, ADAR1 enzyme modifies these endogenous dsRNAs by converting adenosine to inosine, a process that aids in their processing by Dicer and prevents their detection by other dsRNA-sensing proteins [58]. However, in the absence of Dicer, these accumulated dsRNAs can be erroneously identified by dsRNA-sensing innate immune response proteins such as PKR and MDA5, which then activate downstream interferon signaling pathways. This activation results in the production of interferon and the expression of interferon-stimulated genes (ISGs), including IFNβ, thus instigating an antiviral state within the cells [59, 60]. Therefore, Dicer deletion not only leads to the buildup of endogenous dsRNAs and erroneous activation of dsRNA-sensing proteins but also triggers an interferon response and ISG activation, collectively contributing to an antiviral state that hampers viral replication [2]. A deficiency in Dicer can result in reduced levels of miRNAs and disruption in the regulation of miRNA-targeted genes, which affects essential biological processes such as cell differentiation and apoptosis. For instance, research by Witteveldt, et al. found that the absence of miR-673 in Dicer knockout embryonic stem cells led to elevated levels of MAVS and subsequent activation of the interferon response [57].
Utilizing Dicer-knockout mammalian cells as a model for studying antiviral RNAi presents limitations, making it an unsuitable approach for accurately evaluating this mechanism. Instead, several alternative methodologies offer more effective means to assess antiviral RNAi functionality. One such method involves pre-inoculating organisms with viruses that lack VSRs in vivo or employing virus replicons devoid of VSRs in vitro to activate antiviral RNAi pathways. This can be followed by evaluating viral replication rescue through a recombinant virus carrying virus fragments as a reporter system, a technique illustrated in studies by Zhang, et al. and Qiu, et al. [52, 61]. Another approach is to isolate vsiRNAs from infected mammalian tissues using an anti-pan Ago antibody for immunoprecipitation. These vsiRNAs can then be subjected to an in vitro slicing assay with synthetic RNA substrates to test whether Ago proteins, when loaded with vsiRNAs, are capable of cleaving complementary single-stranded RNAs [14]. While AGO2 deficiency results in embryonic lethality in mammals, indicating its crucial role in RNAi, the zebrafish model also reveals limitations due to impaired AGO2 cleavage capacity [2, 62]. Thus, exploring these alternative methods provides more accurate and feasible strategies for studying antiviral RNAi and its functional mechanisms.
Antiviral RNAi Mechanisms and VSR Interactions
Studies on the function of antiviral RNAi in mammals show that different RNA viruses not only cause vsiRNAs to be produced via Dicer, but also encode different dsRNA- binding VSRs to prevent these vsiRNAs from being formed in mammalian cells [13, 61]. Early studies on RNAi caused by synthetic long dsRNA suggested that the Dicer-mediated synthesis of vsiRNAs may be inhibited by the interferon (IFN) response [17]. In both IFN-competent MEFs and Rag1−/− adult mice, who have a full IFN system, Han et al., 2020 demonstrated that NoV RNA replication resulted in the formation of very abundant vsiRNAs when the VSR-B2 protein was missing or made nonfunctional [14]. These vsiRNAs, which were mostly 22 nucleotides long with 2-nt 3′ overhangs, were present in both MEFs and adult mice, suggesting that Dicer processed the viral dsRNA precursors. The findings that Ago2 was not necessary for vsiRNA biogenesis in Dicer-KO MEFs and that vsiRNA synthesis was lacking in these cells provide support for this. Later research revealed that NoVΔB2 or NoVmB2 infection generated vsiRNA-RISC that could regulate Ago2-mediated RNA cleavage in vitro [14]. This cleavage activity requires base pairing between the tenth nucleotide of the vsiRNA and the target RNA. Interestingly, the absence of IFN-I, -II, and -III signaling in Stat1/2−/− mice did not increase RNA slicing by the in vivo-assembled vsiRNA-RISC [14], in contrast to Rag1−/− animals, which have a functioning IFN system. Furthermore, upon infection, there were no discernible variations in Ago2-mediated RNA slicing by endogenous miRNA-RISC between Rag1−/− and Stat1/2−/− mice [14]. These observations diverge from earlier findings indicating that IFN-I signaling inhibits Ago2-mediated RNA slicing in human 293T cells [35].
Han et al., 2020 showed that genetic suppression of RNAi in Ago2-CD MEFs, Dicer-KO and Ago2-KO MEFs, as well as in Ago2-KO MEFs, significantly increased NoV RNA1 replication and RNA3 transcription. This suggests that effective antiviral RNAi requires both Ago2 slicer activity and Dicer-mediated vsiRNA biogenesis [14]. The viral inhibition of RNAi by VSR-B2 in wild-type MEFs also caused an increase in the accumulation of NoV RNA1 and RNA3. In RNAi-defective MEFs, however, VSR-B2’s replication-enhancing impact was negligible, which is consistent with findings in S. cerevisiae missing the RNAi pathway [63]. This implies that the primary function of VSR-B2 is to repress RNAi. Han et al., 2020 also discovered that, independent of VSR-B2 expression, NoV RNA replication in Dicer-KO MEFs was accompanied by a strong activation of the OAS/RNase L system [14]. Viral small RNA populations were present in MEFs and adult mice after extensive NoV RNA replication with functional VSR-B2. These results suggest that, unlike its known suppression of Dicer processing of dsRNA [64, 65], VSR-B2 does not inhibit OAS activation or RNase L-mediated degradation of ssRNAs. It is possible that the VSR-B2-bound long dsRNA remains an effective OAS activator but is poorly recognized by Dicer. Interestingly, the OAS/RNase L system was less activated in Ago2-KO and Ago2-CD MEFs, despite these cells supporting strong replication of NoV RNA1 or R1ΔB2, suggesting that Dicer processing or sequestration of viral dsRNA might attenuate OAS/RNase L activation.
Previous studies have indicated that a variety of wild- type RNA viruses fail to produce a prominent peak of vsiRNAs in commonly used mammalian cell lines or show higher replication levels in human 293T cells following Dicer inactivation. This led to the hypothesis that antiviral RNAi might not significantly inhibit wild-type virus infection in mature cells. Han, et al. demonstrated that RNAi suppression in MEFs significantly enhanced replication of wild-type NoV RNA1, even in the presence of functioning VSR-B2 and resulting in the creation of low-abundance vsiRNAs [14]. This suggests that even in the presence of VSR expression, antiviral RNAi is still somewhat active in MEFs. Ago4 seems to be required for MEFs’ antiviral defense, either by promoting the synthesis of vsiRNA or maintaining the stability of the vsiRNA-RISC [27]. Thus, in comparison to other cell culture methods, MEFs appear to be a more appropriate model for researching antiviral RNAi. Significantly, pan-Argonaute co-immunoprecipitation revealed low-abundance vsiRNAs that may trigger RNA cleavages in pure in vivo vsiRNA-RISC in wild-type NoV-infected adult mice. This shows that the siRNA response has an active antiviral effect against wild- type viral infections.
Given their crucial roles in development, future research should concentrate on creating conditional knockout systems to investigate the in vivo antiviral activities of Dicer or Ago2. However, Han, et al. proposed that RNA interference has a built-in antiviral role in mammals. It was demonstrated that adult mice expressing VSR-B2 inhibited the generation of vsiRNA as well as the activity of vsiRNA-RISC, while having no effect on endogenous miRNAs or the activation of IFN-β and ISGs [14]. Interestingly, VSR-B2 markedly elevated the viral load and produced severe NoV infections in Stat1/2−/− mice with impaired IFN signaling and IFN- competent Rag1−/− animals. However, in both Rag1−/− and Stat1/2−/− mice, NoV mutants missing VSR-B2 did not result in weight loss or illness signs and were mostly eliminated by day 10 post-infection [14]. These results suggest that the RNAi response plays a crucial role in providing protective immunity against viral infections in mammals, irrespective of their IFN response status.
Conclusion
Recent investigations into mammalian antiviral responses have revealed that both positive- and negative- strand RNA viruses can induce antiviral RNAi across a wide variety of cell types, suggesting that this defense mechanism operates more broadly than previously assumed, without being confined to specific viral strains or cellular environments. Many of these viruses produce double-stranded RNA-binding VSRs, which are crucial for maintaining their infectivity, further underscoring the relevance of RNAi in antiviral defense. Notably, research has demonstrated that antiviral RNAi functions independently of the IFN pathway in certain human cancer cell lines, with VSR-B2-mediated suppression of RNAi enhancing the therapeutic efficacy of an oncolytic vesicular stomatitis virus (VSV) variant against these cancer cells. However, contradictory findings indicate that despite the generation of abundant vsiRNAs in human 293T cells, no reduction in influenza A virus (IAV) replication was observed, a finding inconsistent with previous reports. In contrast, primary mouse embryonic fibroblasts (MEFs) have shown effective suppression of IAV replication through antiviral RNAi, highlighting cell-type-specific differences in antiviral RNAi efficacy. While MEFs deficient in the RNAi machinery, such as Ago2D597A MEFs, exhibit increased susceptibility to RNA virus infections, this increased vulnerability is not observed in immortalized AGO2-knockout MEFs, which also lack type I interferon signalling, emphasizing the need for novel infection models that can isolate and better characterize antiviral RNAi functions without interfering with endogenous microRNA activities.
The identification of RNAi as a novel antiviral mechanism in mammals would significantly enhance our understanding of mammalian immunity, offering new therapeutic opportunities. Antiviral RNAi operates as a genetically encoded pathway capable of clearing viral infections without requiring cell death, providing a programmed and infection-specific response. However, key questions remain unresolved, including whether antiviral RNAi is essential in adult mammals, whose antiviral defences are predominantly interferon-dependent. Further therapeutic achievements should be possible if the function of short RNA-mediated antiviral immune systems in mammals is better understood. The SARS-CoV-2 pandemic has sped up the development of mRNA-based treatments and vaccines, which has shown the promise of short RNAs for future therapeutic uses and overcome previous restrictions in nucleic acid medications.
References
-
Napoli C, Lemieux C, Jorgensen R (1990) Introduction of a chimeric Chalcone synthase gene into petunia results in reversible Co-suppression of homologous genes in trans. Plant Cell 2(4): 279-289.
-
Wang J, Li Y (2024) Current advances in antiviral RNA interference in mammals. FEBS J 291(2): 208-216.
-
Fire A, Xu FS, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391(6669): 806-811.
-
Berkhout B (2018) RNAi-mediated antiviral immunity in mammals. Curr Opin Virol 32: 9-14.
-
Song MS, Rossi JJ (2017) Molecular mechanisms of Dicer: endonuclease and enzymatic activity. Biochem J 474(10): 1603-1618.
-
Nakanishi K (2021) Are Argonaute-Associated Tiny RNAs Junk, Inferior miRNAs, or a New Type of Functional RNAs?. Front Mol Biosci 8: 795356.
-
Takahashi T, Ui-Tei K (2020) Mutual regulation of RNA silencing and the IFN response as an antiviral defense system in mammalian cells. Int J Mol Sci 21(4): 1348.
-
Hur S (2019) Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349-375.
-
Tsai K, Courtney DG, Kennedy EM, Cullen BR (2018) Influenza A virus-derived siRNAs increase in the absence of NS1 yet fail to inhibit virus replication. RNA 24(9): 1172-1182.
-
tenOever BR (2017) Questioning antiviral RNAi in mammals. Nat Microbiol 2: 17052.
-
Zhang Y, Xu Y, Dai Y, Li Z, Wang J, et al. (2021) Efficient dicer processing of virus-derived double-stranded RNAs and its modulation by RIG-I-like receptor LGP2. PLoS Pathog 17(8): e1009790.
-
Xu YP, Qiu Y, Zhang B, Chen G, Chen Q, et al. (2019) Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res 29(4): 265-273.
-
Qiu Y, Xu YP, Wang M, Miao M, Zhou H, et al. (2020) Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci Adv 6(6): eaax7989.
-
Han Q, Chen G, Wang J, Jee D, Li WX, et al. (2020) Mechanism and function of antiviral RNA interference in mice. mBio 11(4): e03278-e03319.
-
Takahashi T, Nakano Y, Onomoto K, Yoneyama M, Ui- Tei K (2020) LGP2 virus sensor enhances apoptosis by upregulating apoptosis regulatory genes through TRBP- bound miRNAs during viral infection. Nucleic Acids Res 48(3): 1494-1507.
-
Schuster S, Miesen P, Rij RPV (2019) Antiviral RNAi in insects and mammals: parallels and differences. Viruses 11(5): 448.
-
Maillard PV, Veen AGVD, Poirier EZ, Sousa CRE (2019) Slicing and dicing viruses: antiviral RNA interference in mammals. EMBO J 38(8): e100941.
-
Ma E, MacRae IJ, Kirsch JF, Doudna JA (2008) Autoinhibition of human Dicer by its internal helicase domain. J Mol Biol 380(1): 237-243.
-
Chakravarthy S, Sternberg SH, Kellenberger CA, Doudna JA (2010) Substrate‐specific kinetics of Dicer‐catalyzed RNA processing. J Mol Biol 404(3): 392-402.
-
Kennedy EM, Whisnant AW, Kornepati AVR, Marshall JB, Bogerd HP, et al. (2015) Production of functional small interfering RNAs by an amino‐terminal deletion mutant of human Dicer. Proc Natl Acad Sci USA 112(50): E6945-E6954.
-
Flemr M, Malik R, Franke V, Nejepinska J, Sedlacek R, et al. (2013) A retrotransposon‐driven Dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 155(4): 807-816.
-
Cantini LP, Andino LM, Attaway CC, Butler B, Dumitriu A, et al. (2014) Identification and characterization of Dicer1e, a Dicer1 protein variant, in oral cancer cells. Mol Cancer 13: 190.
-
Hansen SR, Aderounmu AM, Donelick HM, Bass BL (2019) Dicer’s Helicase Domain: A Meeting Place for Regulatory Proteins. Cold Spring Harb Symp Quant Biol 84: 185-193.
-
Taylor DW, Ma E, Shigematsu H, Cianfrocco MA, Noland CL, et al. (2013) Substrate‐specific structural rearrangements of human Dicer. Nat Struct Mol Biol 20(6): 662-670.
-
Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, et al. (2013) ADAR1 forms a complex with Dicer to promote microRNA processing and RNA‐induced gene silencing. Cell 153(3): 575-589.
-
Takahashi T, Heaton SM, Parrish NF (2021) Mammalian antiviral systems directed by small RNA. PLoS Pathog 17(12): e1010091.
-
Adiliaghdam F, Basavappa M, Saunders TL, Harjanto D, Prior JT, et al. (2020) A requirement for Argonaute 4 in mammalian antiviral defense. Cell Rep 30(6): 1690- 1701.
-
Poirier EZ, Buck MD, Chakravarty P, Carvalho J, Frederico B, et al. (2021) An isoform of Dicer protects mammalian stem cells against multiple RNA viruses. Science 373(6551): 231-236.
-
Jeffrey KL (2021) Upping the ante on mammalian antiviral RNA interference. Cell Host Microbe 29: 1333- 1335.
-
Maillard PV, Veen AGVD, Grass SD, Rogers NC, Merits A, et al. (2016) Inactivation of the type I interferon pathway reveals long double‐stranded RNA‐mediated RNA interference in mammalian cells. EMBO J 35(23): 2505-2518.
-
Veen GVD, Maillard PV, Schmidt JM, Lee SA, Grass SD, et al. (2018) The RIG‐I‐like receptor LGP2 inhibits Dicer‐ dependent processing of long double‐stranded RNA and blocks RNA interference in mammalian cells. EMBO J 37(4): e97479.
-
Takahashi T, Nakano Y, Onomoto K, Murakami F, Komori C, et al. (2018) LGP2 virus sensor regulates gene expression network mediated by TRBP‐bound microRNAs. Nucleic Acids Res 46(17): 9134-9147.
-
Komuro A, Homma KY, Negoro T, Barber GN, Horvath CM (2016) The TAR‐RNA binding protein is required for immunoresponses triggered by Cardiovirus infection. Biochem Biophys Res Commun 480(2): 187-193.
-
Girardi E, Lefèvre M, Chane‐Woon‐Ming B, Paro S, Claydon B, et al. (2015) Cross‐species comparative analysis of Dicer proteins during Sindbis virus infection. Sci Rep 5: 10693.
-
Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, et al. (2013) Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14(4): 435-445.
-
Husain B, Mukerji I, Cole JL (2012) Analysis of high‐ affinity binding of protein kinase R to double‐stranded RNA. Biochemistry 51(44): 8764-8770.
-
Xu J, Kong J, Lyu B, Wang X, Qian Q, et al. (2021) The capsid protein of rubella virus antagonizes RNA interference in mammalian cells. Viruses 13(2): 154.
-
Qian Q, Zhou H, Shu T, Mu J, Fang Y, et al. (2020) The capsid protein of Semliki Forest virus antagonizes RNA interference in mammalian cells. J Virol 94(3): e01233-e01319.
-
Li WX, Ding SW (2022) Mammalian viral suppressors of RNA interference. Trends Biochem Sci 47(11): 978-988.
-
Fang Y, Liu Z, Qiu Y, Kong J, Fu Y, et al. (2021) Inhibition of viral suppressor of RNAi proteins by designer peptides protects from enteroviral infection in vivo. Immunity 54(10): 2231-2244.
-
Chen X, Mangala LS, Rodriguez-Aguayo C, Kong X, Lopez-Berestein G, et al. (2018) RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev 37(1): 107-124.
-
Xi HQ, Zhang KC, Li JY, Cui JX, Gao YH, et al. (2017) RNAi-mediated inhibition of Lgr5 leads to decreased angiogenesis in gastric cancer. Oncotarget 8(19): 31581- 31591.
-
Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, et al. (2013) Treatment of HCV infection by targeting microRNA. N Engl J Med 368(18): 1685-1694.
-
Pishavar E, Behravan J (2017) miR-126 as a therapeutic agent for diabetes mellitus. Curr Pharm Des 23(22): 3309-3314.
-
Ding SW, Han Q, Wang J, Li WX (2018) Antiviral RNA interference in mammals. Curr Opin Immunol 54: 109- 114.
-
Wang Q, Wang J, Xu Y, Li Z, Wang B, et al. (2022) The interaction of influenza A NS1 and cellular TRBP protein modulates the function of RNA interference machinery. Front Microbiol 13: 859420.
-
Zhang Y, Li Z, Ye Z, Xu Y, Wang B, et al. (2020) The activation of antiviral RNA interference not only exists in neural progenitor cells but also in somatic cells in mammals. Emerg Microbes Infect 9(1): 1580-1589.
-
Schuster S, Overheul GJ, Bauer L, Kuppeveld FJMV, Rij RPV (2019) No evidence for viral small RNA production and antiviral function of Argonaute 2 in human cells. Sci Rep 9: 13752.
-
Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel MA, et al. (2010) Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog 6(2): e1000764.
-
Girardi E, Chane-Woon-Ming B, Messmer M, Kaukinen P, Pfeffer S (2013) Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus- infected mammalian cells. mBio 4(6): e00698-e00713.
-
Girardi E, Messmer M, Lopez P, Fender A, Chicher J, et al. (2023) Proteomics-based determination of double stranded RNA interactome reveals known and new factors involved in Sindbis virus infection. RNA 29(3): 361-375.
-
Cullen BR (2014) Viruses and RNA interference: issues and controversies. J Virol 88(22): 12934-12936.
-
Witteveldt J, Knol LI, Macias S (2019) MicroRNA-deficient mouse embryonic stem cells acquire a functional interferon response. Elife 8: e44171.
-
Gurung C, Fendereski M, Sapkota K, Guo J, Huang F, et al. (2021) Dicer represses the interferon response and the double-stranded RNA-activated protein kinase pathway in mouse embryonic stem cells. J Biol Chem 296: 100264.
-
Shiromoto Y, Sakurai M, Qu H, Kossenkov AV, Nishikura K (2020) Processing of Alu small RNAs by DICER/ADAR1 complexes and their RNAi targets. RNA 26(12): 1801- 1814.
-
Sadeq S, Al-Hashimi S, Cusack CM, Werner A (2021) Endogenous double-stranded RNA. Noncoding RNA 7(1): 15.
-
Kim S, Ku Y, Ku J, Kim Y (2019) Evidence of aberrant immune response by endogenous double-stranded RNAs: attack from within. Bioessays 41(7): e1900023.
-
Qiu Y, Xu Y, Zhang Y, Zhou H, Deng YQ, et al. (2017) Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 46(6): 992-1004.
-
Chen GR, Sive H, Bartel DP (2017) A seed mismatch enhances Argonaute2-catalyzed cleavage and partially rescues severely impaired cleavage found in fish. Mol Cell 68(6): 1095-1107.
-
Price BD, Eckerle LD, Ball LA, Johnson KL (2005) Nodamura virus RNA replication in Saccharomyces cerevisiae: heterologous gene expression allows replication-dependent colony formation. J Virol 79(1): 495-502.
-
Sullivan CS, Ganem D (2005) A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J Virol 79(12): 7371-7379.
-
Aliyari R, Wu Q, Li HW, Wang XH, Li F, et al. (2008) Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 4(4): 387-397.
-
Li Y, Lu J, Han Y, Fan X, Ding SW (2013) RNA interference functions as an antiviral immunity mechanism in mammals. Science 342(6155): 231-234.
-
Bogerd HP, Skalsky RL, Kennedy EM, Furuse Y, Whisnant AW, et al. (2014) Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J Virol 88(14): 8065-8076.
-
Bartel DP (2018) Metazoan MicroRNAs. Cell 173(1): 20- 51.
- Antifungal Activity of New Acetophenone Derivatives
- Interconnected Microbiomes Human Health Within an Environmental Framework
- Silkworm-Based Vaccine Production for H5N1: A One Health Approach to Pandemic Preparedness
- Microbial Diversity and Lipolytic Activity of Bacteria and Fungi from Oil-Contaminated Sites in Makurdi Metroplois
- Antibiotic Resistance Profile of Bacteria Isolated at the Central Laboratory of the National Hospital Center of Nouakchott
- Epidemiology and Sensitivity to Antibiotics of Germs Isolated from Blood Cultures in the Laboratory of the National Hospital Center of Nouakchott-Mauritania