Systemic Amyloidosis and the Challenge of the Treatment: Clinical Case
A 53-year-old female patient with primary amyloidosis and restrictive cardiomyopathy. For six years the patient has muscle weakness, joint and bone pain, periorbital subcutaneous hemorrhages, bullae on the lower eyelids, macroglossia, gradual thickening of the skin in the shoulder area, body and thighs. Also, there are subcutaneous hemorrhages in other parts of the body. The patient underwent immunofixation of urine and serum and the result showed presence of λ-light chain type and also of IgA. A few months before admission to the clinic, a right ureterolithotomy was performed. After evaluation of the patient, a specific treatment was conducted with melphalan and dehydrocortisone, with a scheme different from the combination regimens used worldwide. The different approach led to good response to the treatment, preserved the renal function and all the cytological tests of the bone marrow did not show plasma cell infiltration. Also, during the treatment, she had no significant proteinuria, no hypoproteinemia, but had persistent pleural effusions, pericardial effusion and persistent edema of the lower extremities.
Introduction
Systemic AL amyloidosis, also called primary amyloidosis, is a disease that occurs as a result of the accumulation of immunoglobulin light chains, which leads to a change in protein structures and they are called amyloids. As a result, insoluble polymer protein fibrils accumulate in the extracellular space of various tissues and organs, leading to organ dysfunction and even death. There are at least 21 different protein precursors that have been identified as causing various types of amyloidosis [1]. The nature of amyloidosis lies in the ability of these proteins to acquire more than one conformation and more than one function, which has earned the name “chameleon proteins” [2].
Amyloidosis affects about 8-9 people per 1,000,000 per year, with the exact number varying according to different authors, but this frequency has remained relatively stable in recent decades. Statistically, 2/3 of the total number of patients are men and only 1% are under the age of 40 [3]. The most commonly affected organs from systemic amyloidosis are the kidneys, heart and liver [4], and the most common manifestations of the disease are massive proteinuria, hypoalbuminemia, dyslipidemia, edema, anasarca, azotemia, macroglossia, hepatosplenomegaly. But all these clinical manifestations are non-specific. Amyloidosis is suspected in patients with at least one of the following four manifestations: nephrotic syndrome with or without renal failure, congestive heart failure due to restrictive cardiomyopathy, hepatomegaly or peripheral neuropathy [5]. In the presence of at least one of the listed clinical manifestations, the performance of immunofixation of serum and urine, as well as subsequent biopsy of subcutaneous adipose tissue is necessary. Usually, when the results are positive from both diagnostic tests, a puncture kidney biopsy is not necessary.
Results
A 53-year-old female patient with complaints of muscle weakness, joint and muscle pain, periorbital subcutaneous hematomas (Figure 1), formation of bullae on the lower eyelids, macroglossia, thickening of the skin on the body , thighs and chest (Figures 2,3), change in the color of the irises and severe swelling of the lower legs. Subcutaneous hematomas are also found in other parts of the body (Figures 4,5). Dystrophic changes of the nails are also observed (Figure 6). Serum and urine immunoelectrophoresis were performed during hospitalization in the clinic and light λ-type chains and serum IgA precipitates were found. The performed electromyography proved bilateral carpal tunnel syndrome.
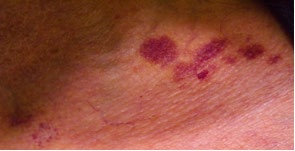
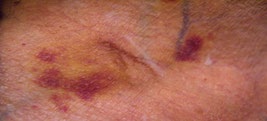
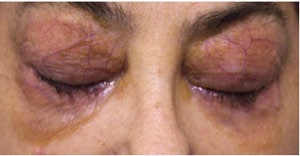
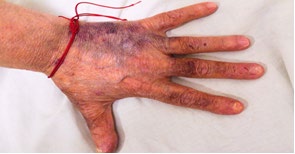
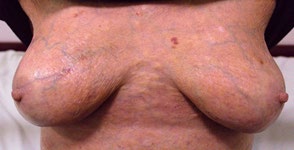
A biopsy of subcutaneous adipose tissue was performed and was stained with Congo red dye and was proven that there was strong evidence of segmental deposition of amyloid-like matter on the basement membrane of sweat glands. Cytological tests of the bone marrow and did not show plasma cell infiltration. Abdominal ultrasound revealed normal renal size and parenchymal echogenicity was slightly increased. Medullary nephrocalcinosis (Figure 7) of non-obstructive type was also described. Few months earlier, before hospitalization in the clinic, was performed ureterolithotomy on the right kidney, because of hydronephrosis in result of a concretion from obstructive type. Also, during the hospitalization were received positive results for HCV-infection, which was referred to a gastroenterologist and a treatment with Harvoni was started for a period of three months. After the successful treatment for HCV-infection, couple of negative results have been reported after antiviral treatment. Also, as a result of hypercalcaemia, intravenous treatment with Zometa was initiated in order to reduce serum calcium levels and prevent future complications of the musculoskeletal system.
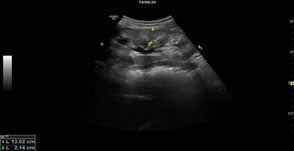
Abdominal echocardiography was performed, which revealed evidence of restrictive cardiomyopathy and the presence of a pericardial effusion of about 100 ml. No pathohistological examination of cardiac tissue was performed. From laboratory tests there was evidence of insignificant proteinuria, lack of hypoproteinemia, the presence of hypercalcemia with slightly increase of the parathyroid hormone. Due to persistent pleural effusions was performed pleural puncture and the pleural puncture was sent for pathological examination. The result was that, there was no evidence of bacterial growth and no tumor cells- transudate.
After а thoroughgoing discussion, it was concluded that edema and the presence of pleural and pericardial effusions were probably associated with systemic AL amyloidosis and possibly amyloid restrictive cardiomyopathy due to the lack of severe hypoproteinemia and proteinuria. Therapy with melphalan, dehydrocortisone and colchicine was started after assessment of the risk group to which the patient belongs. The treatment with melphalan and dehydrocortisone was started with alternating cycles every 4-6 weeks. In our clinic we have more than 10 years of experience in the treatment of a patients with monocyte dyscrasias, especially monoclonal gammopathy with renal significanse-MGRS, by using modified schemes from the standard ones that are used around the world (Table 1). The treatment consists 6-8 cycles according to the following scheme:
| Medicine | Dosage |
| Melphalan | 6-8 mg daily for 5 days |
| Dehydrocortisone | 20 mg daily, on days without Melphalan |
| Colchicine | 1,5-4 mg daily |
Table 1: The drugs used in the treatment 1.
The result of treatment is that we have an improvement of the underlying disease, the renal function remains stable and the risk of myelodysplasia or acute leukemia is much lower. The treatment regimen for the patient from this clinical case was Melphalan 8 mg/daily or 40 mg weekly, plus Dehydrocortisone 20 mg/daily on days without Melphalan. Also, Colchicine was added with dosage of 1.5 to 2 mg/ daily. The total number of immunosuppressive cycles as pathogenetic therapy is 7, for a period of 6 years.
In most patients with primary amyloidosis, type AL, the primary cause of death is progression of restrictive cardiomyopathy or sudden death due to ventricular arrhythmia [6]. Therefore, the condition of the cardiovascular system is one of the important prognostic factors [7]. Due to the high individuality of the disease itself, it is necessary to conduct several routine examinations - echocardiography to assess the cardiovascular system, radiography of flat bones, biopsy of subcutaneous adipose tissue of the abdominal wall, laboratory monitoring of serum calcium, proteinuria, and if necessary cytologic and bone marrow immunoassay. Monitoring of the proteinuria shows the progression of renal involvement in the systemic amyloidosis and it is a prognostic factor for survival. In case of higher protein losses, it is necessary to perform urine immunoelectrophoresis in order to assess the ratio of κ: λ. in patients with nephrotic manifestations the ratio is 1: 5 [8], which shows that λ-light chains predispose to more significant renal infiltration. Surprisingly, however, there is no such relationship between light chain type and renal failure.
Treatment is currently performed according to an immunocytic dyscrasia protocol or monoclonal gammopathy with renal significance, which is close to older regimens for the treatment of primary amyloidosis. Patients receiving such therapy are divided into three groups - low, medium and high risk of complications in therapy. Intermittent doses of melphalan and dexamethasone have been used to reduce the production of aberrant light chains in patients in the medium risk group, as in the clinical case. It is very important to note that patients who have significant cardiac involvement from systematic amyloidosis have a significantly lower survival, regardless of this therapy and it is found difficult to achieve complete remission, but nevertheless- it is not impossible [9, 10].
Treatment Result
During the treatment, the patient maintained stable renal function (Graphic 1), and the established values of the daily proteinuria were always below 0.5 g/l (Graphic 2). No side effects from the applied treatment regimens were registered. At this time the patient is being treated with Colchicine at 2 milligrams daily and 10mg Dehydrocortisone.
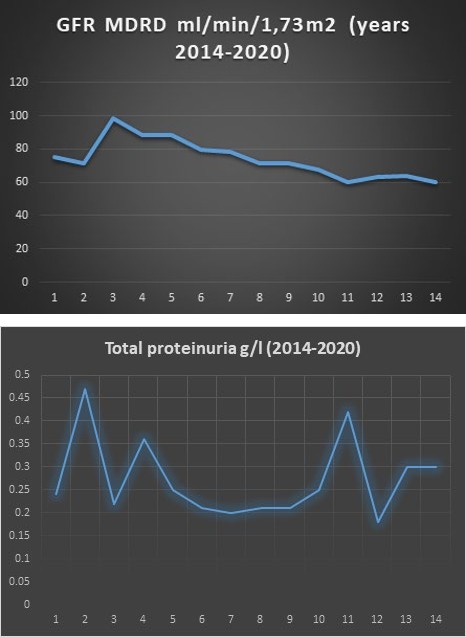
Graphic 1: Kidney function through the years.
Graphic 2: Proteinuria through the years.
Discussion
AL amyloidosis, also called primary amyloidosis, is a clonal proliferative disorder of plasma cells (plasmal dyscrasia) in which fibrils from monoclonal light chains are deposited in the kidneys, heart and other tissues. Affected patients may have AL amyloidosis alone or in combination with other plasma cell dyscrasias (multiple myeloma, Waldenström’s macroglobulinemia, monoclonal gammopathy, etc.). The clinical presentation of AL amyloidosis depends on the nature of the amyloids, the number of the organs affected and which one is the most affected. There are nonspecific systemic symptoms, including fatigue and weight loss, which are common. Other common clinical manifestations include nephrotic syndrome, restrictive cardiomyopathy, peripheral neuropathy, and hepatomegaly with elevated liver enzymes. Other less common but suggestive signs are macroglossia, purpura, and unexplained bleeding from hemorrhagic diathesis. The diagnosis of AL amyloidosis requires a biopsy. Subcutaneous adipose tissue biopsy and bone marrow biopsy are the preferred sites for verification in AL amyloidosis due to the easy access and safety of the methods. AL amyloidosis should be distinguished from other forms of amyloidosis, from localized amyloidosis, and from other types of monoclonal immunoglobulin deposition (MIDD) diseases, as the clinical course and therapy are significantly different.
The survival of the patients with AL amyloidosis depends on the degree of organ involvement, and the median survival is short- from four to six months in those with heart failure. However, with earlier diagnosis, careful patient selection, and the use of currently available chemotherapy regimens, survival can be significantly prolonged [11]. In general, treatment involves the administration of chemotherapy and/ or autologous stem cell transplantation (ASCT) in attempt to suppress the main branch of plasma cells responsible for the formation of AL amyloid [12].
In one of the studies, which were related to the long- term survival of patients with primary amyloidosis on the background of treatment with melphalan and dexamethasone [13], the average survival of about 30 monitored patients in one of the studies was about 90 months. By the first year 51% survived, by the fifth year 16%, and by the tenth - only 4.7% [14]. Although this therapy carries itself a risk of late myelodysplasia or acute leukemia, which will lead to a rapid lethal outcome, especially in elderly patients, it gives a chance to be achieved a remission and to be stopped the progression of the disease. In the described clinical case, we have an evidence of higher survival, than the most patients. Using this quite different approach to the treatment of the patients, from our experience, has showed better results with less side effects. On the threshold of the seventh year from the onset of the disease, there is no better proof for that from a patient feeling well and being in a stable medical condition.
References
-
Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, et al. (2002) Amyloid fibril protein nomenclature-2002. Amyloid 9(3): 197-200.
-
Perutz MF (1997) Amyloid fibrils: mutations make enzyme polymerize. Nature 385(6619): 773-775.
-
DiSalvo TG, King ME, Smith RN (2000) Case 3-2000-A 66-Year-Old Woman with Diabetes, Coronary Disease, Orthostatic Hypotension, and the Nephrotic Syndrome. N Engl J Med 342(4): 264-273.
-
Watanabe T, Saniter T (1975) Morphological and clinical features of renal amyloidosis. Virchows Arch A Pathol Anat Histol 366(2): 125-135.
-
Levinson SS (1991) Kappa/lambda index for confirming urinary free light chain in amyloidosis AL and other plasma cell dyscrasias. Clin Chem 37(6): 1122-1126.
-
Mathew V, Olson LJ, Gertz MA, Hayes DL (1997) Symptomatic conduction system disease in cardiac amyloidosis. Am J Cardiol 80(11): 1491-1492.
-
Dubrey S, Mendes L, Skinner M, Falk RH (1996) Resolution of heart failure in patients with AL amyloidosis. Ann Intern Med 125(6): 481-484.
-
Palmer BF, Alpern RJ (1997) Pathogenesis of edema formation in the nephrotic syndrome. Kidney Int 59: 21- 27.
-
Palladini G, Perfetti V, Obici L, Caccialanza R, Semino A, et al. (2004) Association of melphalan and highdose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood 103(8): 2936-2938.
-
Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, et al. (2007) Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood 110(2): 787-788.
-
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C (2017) Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 135(14): 1357-1377.
-
Gertz MA, Lacy MQ, Gastineau DA, Inwards DJ, Chen MG, et al. (2000) Blood stem cell as therapy for primary systemic amyloidosis (AL). Bone Marrow Transplantation 26: 963-969.
-
Montseny JJ, Kleinknecht D, Meyrier A, Vanhille P, Simon P, et al. (1998) Long-term outcome according to renal histological lesions in 118 patients with monoclonal gammopathies. Nephrol Dial Transplant 13(6): 1438- 1445.
-
Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, et al. (1999) Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood 93(3): 1062- 1066.
- Results of 6-Month Follow-Up of Patients After B-Turp and Thulep
- The Effect of Drinking Water with a High Content of Antimony and Arsenic on the Dynamics of their Distribution in the Kidneys and the Renal Excretory Function in Rats
- Effectiveness and Safety of Tansurethral Thulium Laser Enucleation of the Prostate in the Treatment of BPH: Review
- A Systematic Review on Molecular Pathophysiology Involved in Chronic Kidney Disease and the Role of Animal Models in Drug Discovery to Manage in Chronic Kidney Disease - An Update
- Functional Development of Kidneys in Human Ontogenesis
- Testicular Metastasis: Uncommon Prostate Cancer Case Report